РД 34.17.414-95
ОТРАСЛЕВОЙ РУКОВОДЯЩИЙ ДОКУМЕНТ
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
ПО ХИМИЧЕСКОМУ И ФИЗИКО-ХИМИЧЕСКОМУ ФАЗОВОМУ АНАЛИЗАМ МЕТАЛЛА ЭНЕРГООБОРУДОВАНИЯ ТЕПЛОВЫХ ЭЛЕКТРОСТАНЦИЙ
РД 34.17.414-95
Дата введения 1996-01-01
РАЗРАБОТАНЫ Всероссийским дважды ордена Трудового Красного Знамени теплотехническим научно-исследовательским институтом (АООТ "ВТИ")
ИСПОЛНИТЕЛИ Т.А. Мещерякова
УТВЕРЖДЕНЫ Департаментом науки и техники РАО "EЭС России" 11 сентября 1995 года
Первый заместитель начальника А.П. Берсенев
ВЗАМЕН РД 34.17.414-83
Настоящие Методические указания устанавливают единообразные для всех лабораторий металлов тепловых электростанций методы отбора и подготовки проб, методы проведения анализов, оформления и учета результатов, а также правила хранения проб.
Все методы химического анализа, включенные в настоящие Методические указания, основаны на стандартизированных методах определения основных компонентов и легирующих добавок и отличаются более подробным изложением практических приемов выполнения последовательных операций и отбором необходимых для данного анализа реактивов.
В настоящие Методические указания включены методы анализа, рассчитанные на испытания конкретных марок сталей, применяемых в энергетике, гарантирующие достоверность результатов и, в то же время, наиболее простые в применении на практике.
Все методы построены на использовании стандартных образцов, близких по химическому составу к анализируемым сталям.
Унификация методов анализа имеет целью получение надежных и сопоставимых результатов независимо от места и времени проведения испытаний.
Настоящие Методические указания пригодны для целей сертификации.
Положения настоящего отраслевого нормативного документа подлежат применению расположенными на территории Российской Федерации предприятиями и объединениями предприятий, в том числе союзами, ассоциациями, концернами, акционерными обществами, межотраслевыми, региональными и другими объединениями, имеющими в своем составе (структуре) тепловые электростанции и котельные, независимо от форм собственности и подчинения.
1 ОБЩИЕ ТРЕБОВАНИЯ К ПОДГОТОВКЕ И ПРОВЕДЕНИЮ АНАЛИЗОВ СТАЛИ
1.1 Во всех случаях проведения анализов применяют реактивы квалификации химически чистые (х.ч.) и чистые для анализа (ч.д.а.). При заказе реактивов следует пользоваться каталогом "Химические реактивы и высокочистые вещества" издания 1983 г. и более позднейших лет.
1.2 Для приготовления водных растворов всех реактивов и проведения анализа применяют дистиллированную воду.
1.3 Содержание элемента определяют в 2-х навесках (два определения), взвешенных с точностью до ±0,0002 г. Определения должны выполняться одним испытателем, на одном и том же оборудовании в короткий промежуток времени. Среднее арифметическое значение двух определений принимают за окончательный результат. Расхождение между результатами определений не должно превышать допустимых значений для данного метода анализа и соответствующей концентрации определяемого элемента (приложение 1). В случае превышения допустимых значений анализ повторяют.
1.4 Одновременно в тех же условиях проводят контрольное определение стандартного образца на содержание определяемого элемента.
Стандартный образец - это специальным образом подготовленный материал, в котором с необходимой точностью установлена концентрация всех или части содержавшихся в нем компонентов. Содержание стандартизированных элементов указывается в свидетельстве, прилагаемом к данному стандартному образцу.
Стандартные образцы предназначены для контроля за точностью результатов количественного анализа, проверки метода анализа и средств измерений. Они широко применяются при анализах, основанных на сравнении содержания определяемого элемента в исследуемом образце с известным его значением в стандартом образце.
Стандартный образец подбирается близким к анализируемому металлу по химическому составу и количественному содержанию определяемого элемента. При одном и том же методе анализа всегда берутся одинаковые навески стандартного образца и исследуемого металла.
2 ХИМИЧЕСКИЙ АНАЛИЗ СТАЛЕЙ
2.1 ОТБОР ПРОБ СТАЛИ ДЛЯ АНАЛИЗА (по ГОСТ 7565 и ГОСТ 7122)
2.1.1 Подготовка поверхности и отбор проб
Поверхность металла для отбора проб тщательно очищается от окалины и механических загрязнений. Пробы в одинаковом количестве отбирают в нескольких местах по длине или сечению сверлами до 15 мм или специально заточенным резцом. Твердость сверла или резца должна превышать твердость металла, из которого берут пробы, примерно в 1,5 раза.
Трубы, из которых отбирают пробы, с толщиной стенки 4 мм и менее сверлят по поперечному сечению или по боковой поверхности в нескольких расположенных на одинаковом расстоянии одна от другой точках на одинаковую глубину. Не следует сверлить на всю толщину стенки во избежание попадания в пробу окалины.
Литые или толстостенные детали сверлят по поперечному сечению на расстоянии 1/2 радиуса для круглых деталей или 1/4 диагонали для квадратных деталей, из боковых поверхностей - на 1/2 толщины образца.
Недопустимо попадание на отбираемую пробу масла или охлаждающей эмульсии.
2.1.2 Маркировка и упаковка проб
Для проведения анализа отбирают 20-30 г стружки на чистый лист железа с краями, загнутыми на 8-10 см (использовать бумажную или деревянную подстилки не допускается), и тщательно перемешивают.
Отобранную пробу укладывают в пакет из плотной глянцевой без ворса бумаги (типа кальки). На пакете указывают: наименование ТЭС либо другого предприятия, организации, место отбора пробы, марку стали по сертификату, клеймо и элементы, подлежащие определению, дату отбора. При несоблюдении этих требований анализ не выполняется.
2.2 ОПРЕДЕЛЕНИЕ ОБЩЕГО УГЛЕРОДА
(газообъемный метод по ГОСТ 22536.1)
2.2.1 Сущность метода
Метод основан на сжигании навески стали в токе кислорода при 1250-1350 °C с последующим поглощением образующегося диоксида углерода раствором гидроксида калия (натрия).
Массовую долю углерода определяют по разности первоначального объема газа и объема газа, полученного после поглощения диоксида углерода раствором гидроксида калия (натрия).
2.2.2 Реактивы и приспособления
Калий марганцовокислый, раствор с массовой концентрацией 40 г/дм3 в растворе гидроксида калия (натрия) с массовой концентрацией 400 г/дм3 (для очистки кислорода).
Кислота серная, плотностью 1,84, раствор с массовой концентрацией 20 г/дм3 и разбавленная 4:1.
Калия (натрия) гидроксид, раствор с массовой концентрацией 400 г/дм3 (для заполнения поглотительного сосуда).
Калий двухромовокислый, раствор с массовой концентрацией 150 г/дм3 в серной кислоте, разбавленной 4:1 (для поглощения сернистого газа в промывной склянке).
Натрий хлористый, раствор с массовой концентрацией 260 г/дм3.
Индикатор, метиловый оранжевый, водный раствор с массовой концентрацией 1 г/дм3.
Затворная жидкость: раствор серной кислоты с массовой концентрацией 20 г/дм3 или раствор хлористого натрия (260 г/дм3), подкисленной 2-3 каплями серной кислоты с добавлением в обоих случаях 2-3 капель раствора индикатора метилового оранжевого.
Спирт этиловый.
Ацетон.
Плавни - медь, олово, железо карбонильное, а также оксиды этих металлов.
Плавни перед употреблением сжигают в токе кислорода и проверяют содержание в них углерода, которое не должно превышать величины допускаемых расхождений. На навеску стали в 1 г расходуется 1-1,5 г плавня.
Для проведения анализа используются фарфоровые лодочки неглазированные № 1 и № 2. Перед применением их прокаливают в муфельной печи типа СНОЛ или любого другого типа при температуре не ниже 900 °С в течение одного рабочего дня (7-8 часов). Лодочки хранят в эксикаторе, шлиф крышки которого не покрывают смазкой во избежание попадания смазки в пробу и искажения результатов анализа. При необходимости перед анализом лодочки прокаливают в токе кислорода при рабочей температуре в течение 3-5 мин.
2.2.3 Аппаратура
Установка для газообъемного определения углерода состоит из следующих элементов (рисунок 1): баллона с кислородом или кислородопровода, снабженного редукционными вентилями для пуска и регулирования тока кислорода 1; промывной склянки Тищенко, наполненной раствором марганцовокислого калия с массовой концентрацией 40 г/дм3 в растворе гидроксида калия (натрия) с массовой концентрацией 400 г/дм3 2; промывной склянки, наполненной концентрированной серной кислотой для поглощения паров воды 3; стеклянного крана, с помощью которого регулируют скорость тока кислорода 4; трубчатой горизонтальной печи с силитовыми нагревателями 5; металлического кожуха, в который заключена печь 6; терморегулятора типа ПСР или другого подобного типа 7; регулятора напряжения типа PHО-250-10 или другого подобного типа 8; фарфоровой или огнеупорной муллитокремнеземистой трубки длиной 750 мм, внутренним диаметром 18-22 мм, концы которой должны выступать из печи не менее, чем на 160 мм с каждой стороны 9; лодочки фарфоровой 10; стеклянной трубки с расширением, заполненной ватой для удержания твердых оксидов, уносимых из печи током кислорода 11; промывной склянки с раствором двухромовокислого калия в серной кислоте 12; газоанализатора ГОУ-1 или КГА-4, включающего змеевиковый холодильник для охлаждения поступающей из печи газовой смеси (диоксида углерода и кислорода) 13; двухходового крана, через который газовая смесь поступает в эвдиометр (газо-измерительную бюретку) 14; одноходового крана, соединяющего эвдиометр с атмосферой 15; эвдиометра, к узкой части которого прикреплена градуированная на процентное содержание углерода подвижная шкала 16; термометра для измерения температуры газов 17; сосуда для поглощения диоксида углерода, наполненного раствором гидроксида калия (натрия) 18; уравнительной склянки, наполненной затворной жидкостью 19.
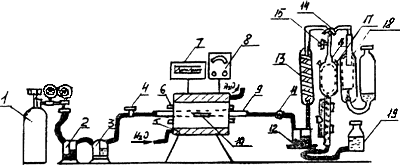
Рисунок 1
2.2.4 Подготовка установки к анализу
Установку (см. рисунок 1) приводят в рабочее состояние: концы фарфоровой трубки 9 закрывают хорошо подогнанными резиновыми пробками, в которые вставлены стеклянные трубки для подсоединения резиновых шлангов.
Эвдиометр 16 и поглотительный сосуд 18 заполняют соответствующими растворами так, чтобы их поплавки закрывали входные отверстия.
Холодильник 13 и кожух эвдиометра 16 заполняют водой. Собрав установку, нагревают печь 5 до рабочей температуры и проверяют на герметичность фарфоровую трубку, все соединения и краны. Для этого по системе пропускают кислород в течение 1-2 мин, затем перекрывают его краном 4, а краном 14 соединяют печь с эвдиометром, уравнительная склянка 19 устанавливается на уровне нуля шкалы измерения.
Если уровень жидкости в эвдиометре, понизившись, сохраняется неизменным в течение 2-3 мин, герметичность системы можно считать приемлемой. Если же уровень непрерывно повышается, проверяют герметичность каждого соединения, последовательно перекрывая его соответствующим краном или пережимом соединительных шлангов.
Если установка негерметична, но при проверке отдельных соединений не обнаружено место утечки, установку разбирают, тщательно протирают все краны, смазывают вакуумной смазкой и проверяют снова.
Эвдиометр периодически промывают теплым хромовым раствором. Вату в расширении стеклянной трубки 11 меняют по мере загрязнения.
2.2.5 Проведение анализа
Для определения углерода при содержании его в стали (по сертификату) до 0,2% применяют микроэвдиометр, шкала бюретки которого рассчитана на содержание углерода до 0,25%; при более высоком содержании углерода применяют эвдиометр со шкалой, калиброванной до 1,5%. Содержание углерода в количестве 1,5-2,0% определяют эвдиометром со шкалой, рассчитанной до 4% (в отсутствии такой бюретки можно пользоваться эвдиометром со шкалой до 1,5%, соответственно уменьшив при этом массу навески образца).
Перед началом работы через фарфоровую трубку 9, нагретую до температуры 1250-1350 °C, пропускают кислород в течение 3-5 мин. В начале анализа и через каждые 2-3 ч работы сжигают 2-3 навески стандартного образца для проверки работы установки.
Навеску стали массой 1 г в виде стружки толщиной не более 3 мм (анализ стружки с цветом побежалости не допускается), предварительно промытую этиловым спиртом или ацетоном и высушенную, помещают в лодочку 10, прибавляют сверху равномерным слоем 1-1,5 г плавня. Лодочку 10 с помощью специального крючка задвигают по направлению движения кислорода в наиболее накаленную часть трубки 9, которую сразу же закрывают резиновой пробкой. Через 10-20 с начинают пропускать кислород через склянки 2 и 3 со скоростью пропускания 0,2-0,4 дм3/мин. Затем вводят газовую смесь в эвдиометр при помощи крана 14 до тех пор, пока жидкость в нем не опустится до нулевого деления, после чего кран закрывают. Сгорание стружки и заполнение эвдиометра происходят в течение 5-7 мин. Затем прекращают подачу кислорода краном 4 перед печью и краном 14 перед эвдиометром; открывают фарфоровую трубку и извлекают лодочку. При полном сгорании плав равномерно распределен по дну лодочки, нет несгоревших кусков стали и плавня, нет коричневого налета на стенках лодочки. В противном случае следует взять новую навеску и повторить определение.
Затем измеряют объем смеси газов. Для этого уравнительную склянку 19 перемещают вдоль эвдиометра, находят положение, при котором уровни жидкостей в селянке и эвдиометре окажутся одинаковыми, и устанавливают нулевое деление подвижной шкалы. После измерения общего объема смеси газов краном 14 соединяют эвдиометр с поглотительным сосудом 18 и перекачивают газовую смесь через раствор щелочи, находящейся в поглотительном сосуде, и обратно в эвдиометр путем поднимания и опускания уравнительной склянки. Операцию перекачивания смеси газов повторяют дважды.
Необходимо следить за тем, чтобы в эвдиометре не осталось пузырьков газа, а при обратной перегонке газа в эвдиометр поплавок поглотительного сосуда плотно закрывал вход в нее.
После выдержки в течение 20 с измеряют объем поглощенного газа: установив жидкость в эвдиометре и уравнительной склянке на одном уровне, записывают показания шкалы. Измеряют через каждые 1-1,5 ч температуру газа в эвдиометре и атмосферное давление. В полученный результат вводят поправку на температуру и атмосферное давление в соответствие с таблицей, приведенной в приложении к описанию газоанализатора.
2.2.6 Расчет результатов анализа
Содержание углерода (C) в процентах вычисляют по формуле
, (1)
где a - показание шкалы эвдиометра, соответствующее процентному содержанию углерода в пробе;
к - поправочный коэффициент на температуру и атмосферное давление (из таблицы);
mисл - масса навески исследуемого образца, г.
При наличии углерода в плавне проводят контрольный опыт, и содержание углерода в этом случае определяется по формуле
, (2)
где а1 - показание шкалы эвдиометра, соответствующее процентному содержанию углерода в плавне.
2.3 ОПРЕДЕЛЕНИЕ ОБЩЕГО УГЛЕРОДА
(кулонометрический метод по ГОСТ 22536.1)
2.3.1 Сущность метода
Метод основан на сжигании навески стали в токе кислорода при 1250-1350 °C с последующим поглощением образовавшегося диоксида углерода поглотительным раствором с определенным начальным значением рН и последующем измерении на установке для кулонометрического титрования, необходимого для восстановления исходного значения рН количества электричества, которое пропорционально массовой доле углерода в навеске стали. Выбор метода (по пп.2.2 или 2.3) зависит от наличия прибора.
2.3.2 Реактивы и приспособления
Поглотительный и вспомогательный растворы готовятся в соответствии с типом применяемой кулонометрической установки.
Гидроперит.
Спирт этиловый.
Ацетон.
Свинец гранулированный.
Плавни - медь, олово, железо карбонильное, а также оксиды этих металлов. Перед применением их проверяют на содержание углерода, которое не должно превышать величины допускаемых расхождений.
Трубки фарфоровые или огнеупорные муллитокремнеземистые длиной 600-800 мм, внутренним диаметром 20-22 мм.
Лодочки фарфоровые неглазированные № 1 или № 2 (см. п.2.2.2).
2.3.3 Аппаратура
Кулонометрическая установка типа АН 7529, АН 7560 со всеми принадлежностями (кулонометр, поглотительные сосуды, рН-метр, корректор массы) или любого другого типа, обеспечивающая необходимую точность результатов анализа.
Горизонтальная трубчатая печь любого типа, обеспечивающая нагрев до температуры 1400 °С.
2.3.4 Подготовка к анализу
Прибор подготавливают к работе в соответствии с инструкцией и проводят настройку анализатора, которая включает следующие операции, выполняемые последовательно: 1) проверка и компенсация "холостого счета"; 2) проверка и регулировка чувствительности прибора; 3) градуировка по стандартным образцам. Перед началом работы прокаливают фарфоровую трубку и пропускают кислород, пока показания прибора не станут соответствовать показаниям "холостого счета".
Для контроля правильности результатов анализа перед началом работы сжигают не менее трех навесок стандартного образца стали с известной массовой долей углерода и близкой к определяемой, и через каждые 3 ч повторяют контроль.
2.3.5 Проведение анализа
Навеску стали (стружку) массой 0,5 г помещают в фарфоровую лодочку и покрывают слоем плавня, помещают в наиболее нагретую часть печи и сжигают в токе кислорода до тех пор, пока показания прибора изменяются на величину "холостого счета". Как правило, это происходит в течение 2,5-3,5 мин.
2.3.6 Обработка результатов
Массовую долю углерода определяют по цифровому табло анализатора за вычетом поправок, которые образуются, если исследуются стали с различным содержанием углерода.
2.4 ОПРЕДЕЛЕНИЕ СВОБОДНОГО УГЛЕРОДА
(по ГОСТ 22536.1)
2.4.1 Сущность метода
Графит и углерод отжига представляют собой формы так называемого свободного (элементарного) углерода, встречающегося в чугунах и графитизированных сталях.
Углерод отжига является продуктом распада цементита и образуется при длительном тепловом воздействии на сталь. Обе формы свободного углерода - графит (кристаллическая форма) и углерод отжига (аморфная форма) - практически не растворяются при воздействии на них кислот, в частности разбавленной (1:1, 1:2, 1:3) азотной кислоте, в которой растворяются карбиды. При растворении стали в разбавленной азотной кислоте графит и углерод отжига окажутся совместно в нерастворимом осадке, который отфильтровывают на асбест, высушивают и определяют массовую долю свободного углерода газообъемным или кулонометрическим методом.
При определении свободного углерода особое внимание уделяется отбору проб стали. Для определения свободного углерода пробу следует брать в виде кусочков размером 3-4 мм, так как при сверлении может произойти выкрашивание графита, что приведет к искажению результатов. Если же образец приходятся сверлить, то используют для навески всю сталь, включая мелочь.
2.4.2 Реактивы
Кислота азотная, разбавленная 1:2.
Кислота фтористоводородная, плотностью 1,12.
Кислота соляная, плотностью 1,19.
Асбест мелковолокнистый.
Асбест несколько раз обрабатывают соляной кислотой при кипячении, пока она не перестанет окрашиваться солями железа в желтый цвет, промывают несколько раз горячей водой до отрицательной реакции на хлор-ион. Промытый асбест сушат и прокаливают при температуре 800-850 °С в течение 0,5-1,0 ч. Хранят асбест в закрытом сосуде.
Для внесения поправки на содержание углерода в асбесте сжигают его в печи в токе кислорода в таком же количестве, как и при проведении анализа. Полученное в нем количество углерода вычитают из результатов определения свободного углерода в анализируемой пробе.
2.4.3 Проведение анализа
Навеску массой 0,5-2,0 г растворяют в стакане вместимостью 200 см3 в 50 см3 азотной кислоты без нагревания. После прекращения растворения в течение 2 ч выдерживают при слабом нагревании. Растворение навески считается законченным, если при перемешивании раствора имеющийся в нем темный нерастворимый осадок поднимается кверху и затем медленно опускается на дно стакана. При длительном нагревании осадок растворяется.
При наличии в растворе хлопьевидного осадка кремниевой кислоты к горячему раствору осторожно по каплям приливают раствор фтористоводородной кислоты по 0,5 см3 на 1 г навески и кипятят 8-10 мин для образования растворимой кремнефтористоводородной кислоты. Содержимое стакана разбавляют горячей водой до 100 см3.
Горячий раствор отфильтровывают на слой прокаленного асбеста, вложенный в воронку, и хорошо промывают горячей водой, подкисленной азотной кислотой, а потом - несколько раз только водой. Осадок графита вместе с асбестом переносят пинцетом в прокаленную фарфоровую лодочку, высушивают и определяют массовую долю свободного углерода газообъемным или кулонометрическим методом. Плавень в данном случае не применяется.
2.4.4 Расчет результатов
Содержание свободного углерода вычисляется так же, как и при определении содержания общего углерода. В полученный результат следует внести поправку на содержание углерода в асбесте.
2.5 ОПРЕДЕЛЕНИЕ СЕРЫ ЙОДОМЕТРИЧЕСКИМ МЕТОДОМ
(по ГОСТ 12345)
2.5.1 Сущность метода
При сжигании навески стали с плавнем в токе кислорода при температуре не ниже 1250-1300 °С сера выгорает и в виде сернистого газа выносится током кислорода в поглотительный сосуд с крахмальным водным раствором. При соприкосновении с водой сернистый газ образует сернистую кислоту, которую окисляют до серной кислоты титрованным раствором йода.
2.5.2 Реактивы и приспособления
Плавни - медь, олово, а также оксиды этих металлов предварительно проверяют на содержание серы в условиях, при которых проводят анализ.
Крахмал, раствор с массовой концентрацией 10 г/дм3; для приготовления 100 г раствора - 1 г крахмала растворяют в 20 см3 воды, вливают в 80 см3 кипящей воды, кипятят 5 мин и охлаждают.
Йод кристаллический; для приготовления титрованного раствора 1,9845 г йода взвешивают в стаканчике с притертой пробкой, пересыпают в колбу, содержащую 15 г йодистого калия и 60 см3 воды, колбу закрывают пришлифованной пробкой. После растворения йода раствор переливают в склянку из темного стекла, разбавляют водой до 5 дм3 и хорошо перемешивают.
Титр раствора йода устанавливают по стандартному образцу с известным содержанием серы. Сжигание стандартного образца проводят в тех же условиях, что и сам анализ.
Титр раствора йода (T) в граммах в кубическом сантиметре вычисляют по формуле
, (3)
где C - содержание серы в стандартом образце, %;
mст - навеска стандартного образца, г;
V - объем раствора йода, пошедшего на титрование, см3.
Раствор йода можно приготовить из 0,01-нормального фиксонала йода. Для этого содержимое ампулы растворяют в 1 дм3 воды, отбирают 330 см3 раствора и разбавляют его до 1 дм3 водой. Раствор йода следует хранить в плотно закрытой темной банке. Титр раствора йода проверяют при каждом определении содержания серы.
Фарфоровые лодочки №№ 1 и 2, прокаленные в токе кислорода при температуре 1250-1300 °С в течение 3-7 мин.
2.5.3 Аппаратура
Установка для определения серы методом сжигания (рисунок 2) состоит из следующих основных элементов: баллона с кислородом или кислородопровода, снабженного редукционными вентилями для пуска и регулирования тока кислорода 1; поглотительных склянок 2, 3 для очистки кислорода, поступающего в печь (склянка 2 заполнена раствором марганцовокислого калия с массовой концентрацией 40 г/дм3, склянка 3 - сухим хлористым кальцием, поверх которого имеется прослойка из стеклянной ваты, а на ней слой натронной извести); крана 4 для регулирования скорости поступления кислорода в трубчатую печь 5 с силитовыми стержнями, допускающими нагрев до температуры не ниже 1450 °С; фарфоровой неглазурованной трубки 6 внутренним диаметром 20-22 мм, концы которой должны выступать из печи на 160 мм (трубку перед работой прокаливают при температуре 1250-1300 °С в атмосфере кислорода); стеклянного шара 7, наполненного ватой; поглотительного аппарата 8, состоящего из двух одинаковых сосудов диаметром 30-35 мм и высотой 150 мм, в нижней части которых имеются краны для слива жидкости по окончании анализа (в левый сосуд входит барботер 9, через который проходит смесь сернистого газа и кислорода; распыление газов через барботер улучшает поглощение сернистого газа водой, в правом сосуде во время титрования находится раствор - индикатор окраски, и бюретки 10 с титрованным раствором йода.
2.5.4 Подготовка установки к анализу
Фарфоровую трубку 6 (см. рисунок 2) необходимо проверить на герметичность, для чего по достижении в печи температуры 1250-1300 °С закрывают трубку с обеих сторон пробками, в которые вставлены стеклянные трубки с резиновыми шлангами, наливают в поглотительный аппарат 70-75 см3 воды и 5 см3 крахмала, приливают из бюретки три-пять капель раствора йода до появления слабо-голубой окраски и, открыв кран 4, пропускают кислород в сосуд 8. Обесцвечивание раствора в поглотительном сосуде указывает на выделение из трубки восстановительных газообразных веществ, реагирующих с йодом. В этом случае следует, не прекращая подачи кислорода, приливать по каплям раствор йода из бюретки в поглотительный сосуд до получения устойчивой голубой окраски. Работу установки проверяют сжиганием двух-трех навесок стандартного образца.
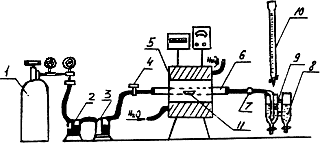
Рисунок 2
2.5.5 Проведение анализа
Навеску стали (или стандартного образца) массой 0,5-1 г помещают в фарфоровую лодочку 11, покрывают сверху равномерным слоем плавня массой 0,5-1 г. Лодочку с помощью специального крючка задвигают по направлению подачи кислорода в наиболее нагретую часть фарфоровой трубки 6, которую плотно закрывают резиновой пробкой и выдерживают в течение 0,5 мин без доступа кислорода. Уровень жидкости в отводящей трубке с барботером 9 в этих условиях поднимается вверх; при этом необходимо следить, чтобы жидкость не затянуло в шар 7, наполненный ватой. Открывают кран 4, подводящий кислород, и пропускают последний со скоростью 2,5 дм3/мин. Смесь газов, поступающая из печи в поглотительный сосуд, обесцвечивает раствор. Из бюретки осторожно по каплям приливают титрованный раствор йода так, чтобы раствор в поглотительном сосуде все время оставался окрашенным в первоначальный бледно-голубой цвет. По окончании процесса обесцвечивания кислород пропускают еще в течение 1 мин.
Лодочку 11 вынимают из печи и выливают раствор из поглотительного сосуда. При полном выгорании плав равномерно расположен по дну лодочки плотной массой, коричневых окислов по стенкам не должно быть.
Анализ стандартного образца периодически повторяют.
2.5.6 Расчет результатов анализа
Содержание серы (S) в процентах определяют по формуле
, (4)
где V - объем раствора йода, израсходованного на титрование, см3;
T - титр раствора йода, г/см3;
mисл - навеска исследуемого образца, г.
Примечания: 1. Заниженные результаты могут получиться из-за использования зашлакованной трубки 6 или образования пузырей в лодочке во время сплавления, в которых задерживается сернистый газ.
2. Завышенные результаты могут получиться при недостаточном предварительном прокаливании трубки 6 и лодочки или при обгорании пробок, закрывающих трубку 6.
2.6 ОПРЕДЕЛЕНИЕ КРЕМНИЯ ГРАВИМЕТРИЧЕСКИМ МЕТОДОМ
(по ГОСТ 12346)
2.6.1 Сущность метода
В сталях и сплавах кремний в основном находится в виде силицидов железа. При обработке стали кислотами силициды разлагаются и переходят в кремниевую кислоту.
Наиболее распространенным методом определения содержания кремния является гравиметрический, основанный на растворении стали в кислотах с образованием кремниевой кислоты в виде коллоидного раствора.
После выпаривания раствора кремниевая кислота обезвоживается и из коллоидного раствора переходит в нерастворимое состояние. При разложении кислотами проб легированных сталей вместе с кремниевой кислотой в нерастворимый осадок переходят вольфрамовая кислота, пятиокись ниобия и частично диоксид титана. В этом случае осадок после взвешивания обрабатывают серной и фтористо-водородной кислотами и вторично прокаливают.
2.6.2 Реактивы
Кислота серная, пл. 1,84; 1:4.
Кислота соляная, пл. 1.19; 5:95.
Кислота азотная, пл. 1,4.
Аммоний роданистый, раствор с массовой концентрацией 50 г/дм3.
Кислота фтористоводородная, пл. 1,12.
2.6.3 Проведение анализа
Для углеродистых, низко- и среднелегированных сталей навеску (стружку) массой 0,5-2 г растворяют в стакане вместимостью 250-300 см3 в 50 см3 серной кислоты 1:4. Когда стружка растворится, осторожно по каплям прибавляют азотную кислоту пл. 1,4 до прекращения выделения оксида азота. После окисления раствор выпаривают до выделения белых паров серной кислоты. Содержимое стакана охлаждают, осторожно приливают 10 см3 соляной кислоты пл. 1,19, нагревают, прибавляют 100 см3 горячей дистиллированной воды и снова нагревают в течение 10 мин при температуре 50-70 °С до растворения солей.
Осадок кремнекислоты отфильтровывают на плотный фильтр "синяя лента", промывают горячей разбавленной (5:95) соляной кислотой до отрицательной реакции на железо (проба с раствором роданистого аммония), затем несколько раз промывают горячей водой. Полученный осадок вместе с фильтром помещают в платиновый тигель, подсушивают, сжигают и прокаливают в муфеле при температуре - 1000-1050 °C. Тигель с осадком диоксида кремния охлаждают и взвешивают. Если осадок диоксида кремния загрязнен вольфрамом, кислотой, пятиокисью ниобия, оксидами хрома, никеля, железа и других элементов, его смачивают двумя-тремя каплями воды, прибавляют пять-восемь капель серной кислоты пл. 1,84 и 1-2 см3 10%-ной фтористоводородной кислоты. Содержание тигля осторожно выпаривают досуха. Сухой остаток прокаливают при температуре 1000-1050 °C. Тигель с осадком охлаждают в эксикаторе и взвешивают.
2.6.4 Расчет результатов анализа
Содержание кремния (Si) в процентах определяют по формуле
, (5)
где А - масса тигля с осадком диоксида кремния до обработки серной и фтористоводородной кислотами, г;
Б - масса тигля с осадком после обработки серной и фтористоводородной кислотами, г;
0,4674 - коэффициент пересчета диоксида кремния на кремний;
mисл - навеска исследуемого образца, г.
Примечание. В присутствии вольфрама осадок прокаливают при температуре не выше 800 °C во избежание улетучивания оксида вольфрама, что может дать завышенные показателя содержания кремния в стали.
2.7 ОПРЕДЕЛЕНИЕ КРЕМНИЯ ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
(по ГОСТ 22536.4)
2.7.1 Сущность метода
Кремнекислота, находящаяся в растворе, образует с молибденовокислым аммонием кремнемолибденовую гетерополикислоту желтого цвета, ее восстанавливают тиомочевиной в присутствии сернокислой меди. Молибденовая синь представляет собой стойкое соединение и дает возможность определить кремний в высоколегированных сталях в присутствии всех компонентов, в частности вольфрама и ванадия.
Выбор метода (по пп.2.6 для 2.7) зависит от наличия прибора.
2.7.2 Реактивы
Кислота серная, разбавленная 1:8.
Кислота азотная, пл. 1,4.
Аммоний молибденовокислый, перекристаллизованный из спирта, раствор с массовой концентрацией 50 г/дм3.
Медь сернокислая, раствор с массовой концентрацией 4 г/дм3 в серной кислоте, разбавленной 1:3.
Тиомочевина, раствор с массовой концентрацией 100 г/дм3.
2.7.3 Проведение анализа
Навеску стали (стружку) массой 0,1-0,2 г растворяют в 15 см3 серной кислоты (1:8) в конической колбе вместимостью 100 см3. Растворение ведут при слабом нагревании, не доводя до кипения. После полного растворения навески прибавляют по каплям азотную кислоту до прекращения выделения оксида азота, раствор охлаждают до комнатной температуры и переводят в мерную колбу вместимостью 100 см3, доводят до метки водой.
На анализ отбирают по 5-10 см3 прозрачного раствора (если сталь содержит вольфрам или ниобий, отфильтровывают часть раствора) в мерную колбу вместимостью 100 см3, прибавляют 10 см3 раствора молибденовокислого аммония, перемешивают и до полного развития желтой окраски раствору дают отстояться 15 мин. Затем добавляют 10 см3 раствора сернокислой меди, перемешивают, добавляют 5 см3 раствора тиомочевины, доливают до метки водой и перемешивают. Дают отстояться 5 мин до развития синей окраски.
Полученную синюю окраску измеряют на фотоколориметре с красным светофильтром в кювете рабочей длиной 30 мм. В качестве холостой пробы используют анализируемый раствор со всеми реактивами, кроме молибденовокислого аммония.
Содержание кремния находят методом сравнения оптической плотности испытуемого и стандартного растворов. Стандартный раствор готовят из стандартного образца, близкого по химическому составу к исследуемому, и проводят через весь ход анализа.
2.7.4 Расчет результатов анализа
Содержание кремния (Si) в процентах
, (6)
где C - содержание кремния в стандартном образце, %;
Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора;
mст - навеска стандартного образца, соответствующая аликвотной части, г;
mисл - навеска исследуемого образца, соответствующая аликвотной части, г.
При одинаковых навесках стандартного и исследуемого образцов, взятых для анализа, mст и mисл не учитываются.
2.8 ОПРЕДЕЛЕНИЕ ФОСФОРА ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ В УГЛЕРОДИСТЫХ И НИЗКОЛЕГИРОВАННЫХ СТАЛЯХ
(по ГОСТ 22536.3)
2.8.1 Сущность метода
В кислой среде фосфорная кислота образует с молибденовокислым аммонием желтую фосфорномолибденовую кислоту, которая восстанавливается тиомочевиной в присутствии сернокислой меди до синего комплексного соединения.
2.8.2 Реактивы
Азотная кислота, разбавленная 1:1.
Соляная кислота, пл. 1,19 и пл. 1,105.
Калий марганцовокислый, раствор с массовой концентрацией 40 г/дм3.
Натрий азотистокислый, раствор с массовой концентрацией 50 г/дм3.
Аммиак водный, разбавленный 1:1.
Аммоний молибденовокислый, перекристаллизованный, раствор с массовой концентрацией 50 г/дм3.
Восстановительная смесь: 150 см3 раствора сернокислой меди с массовой концентрацией 10 г/дм3 смешивают с 700 см3 раствора тиомочевины с массовой концентрацией 80 г/дм3. После отстаивания в течение 24 ч смесь фильтруют через "синий" фильтр, осадок отбрасывают.
2.8.3 Проведение анализа
Навеску стали (стружку) массой 1 г растворяют при нагревании в 20-30 см3 горячей азотной кислоте (1:1) в стакане вместимостью 200 см3. После растворения навески прибавляют по каплям раствор марганцовокислого калия до выпадения бурого осадка диоксида марганца и кипятят 2-3 мин. Затем так же по каплям прибавляют раствор азотистокислого натрия до просветления раствора и избыток 2-3 капли, и кипятят до полного удаления оксидов азота.
Раствор выпаривают досуха, прибавляют 10 см3 соляной кислоты (пл. 1,19) и опять выпаривают досуха. Затем приливают 15 см3 соляной кислоты (пл. 1,19), растворяют при нагревании соли, прибавляют 20-30 см3 воды, охлаждают и отфильтровывают раствор от кремниевой кислоты в мерную колбу вместимостью 100 см3, доливают раствор до метки и перемешивают.
На анализ отбирают 10 см3 раствора в мерную колбу вместимостью 100 см3, прибавляют 15 см3 воды и нейтрализуют по каплям раствором водного аммиака (1:1) до начала выпадения гидроксида железа, который растворяют по каплям раствором соляной кислоты (пл. 1,105) и 2 см3 в избыток. Затем прибавляют 10 см3 восстановительной смеси (раствор обесцвечивается), через 1-2 мин прибавляют 10 см3 раствора соляной кислоты (пл. 1,105) и по каплям, непрерывно перемешивая, 8 см3 раствора молибденовокислого аммония. Раствор перемешивают в течение 1-2 мин, потом разбавляют водой до метки и перемешивают.
Через 10 мин измеряют оптическую плотность на фотоколориметре с красным светофильтром в кюветах рабочей длиной 50 мм. В качестве холостой пробы используют анализируемый раствор со всеми реактивами, кроме молибденовокислого аммония.
Параллельно проводят анализ стандартного образца, близкого по химическому составу к исследуемому.
2.8.4 Расчет результатов анализа
Содержание фосфора (P) в процентах вычисляют по формуле
, (7)
где C - содержание фосфора в стандартном образце, %;
Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора.
2.9 ОПРЕДЕЛЕНИЕ ФОСФОРА ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ В СТАЛЯХ С СОДЕРЖАНИЕМ ТИТАНА, НИОБИЯ, ВОЛЬФРАМА
(по ГОСТ 12347)
2.9.1 Сущность метода
В кислой среде фосфорная кислота образует с молибдатом аммония желтую фосфорномолибденовую гетерополикислоту. При восстановлении молибдена, входящего в ее состав (комплекс), до пятивалентного, раствор окрашивается в синий цвет, фосфор определяют без отделения компонентов сплава. Титан, ниобий, ванадий и вольфрам (частично) связываются в комплекс фтористоводородной кислотой. Избыток фтористоводородной кислоты удаляют борной кислотой, так как свободная фтористоводородная кислота увеличивает интенсивность синей окраски и мешает определению содержания фосфора.
2.9.2 Реактивы
Смесь кислот: 3 части соляной кислоты пл. 1,19 и 1 часть азотной кислоты, пл. 1,4.
Кислота фтористоводородная, пл. 1,12.
Кислота борная.
Аммиак водный, разбавленный 1:1.
Кислота серная, разбавленная 1:5.
Соль закиси железа и аммония двойная сернокислая (соль Мора), раствор с массовой концентрацией 100 г/дм3 в растворе серной кислоты с массовой концентрацией 50 г/дм3.
Перекись водорода, раствор с массовой концентрацией 30 г/дм3.
Медь сернокислая, раствор с массовой концентрацией 10 г/дм3.
Тиомочевина, раствор с массовой концентрацией 100 г/дм3.
Аммоний молибденовокислый, раствор с массовой концентрацией 50 г/дм3.
2.9.3 Проведение анализа
Навеску стали (стружку) массой 1 г растворяют в 30 см3 смеси кислот в конической колбе вместимостью 250 см3. Колбу накрывают часовым стеклом и следят за тем, чтобы раствор не выпаривался. Растворение ведется не менее 45 мин и не более 2 ч. Затем раствор охлаждают, добавляют 15-20 см3 воды и оставляют на 24 ч (особенно если в стали присутствуют вольфрам и ниобий - для более полного их гидролиза). Затем на холоду приливают резиновой пипеткой 1 см3 фтористоводородной кислоты, перемешивают 10 с; приливают еще 2 см3 фтористоводородной кислоты, перемешивают 10 с; приливают 5 см3 раствора перекиси водорода, перемешивают 10 с; прибавляют 3 г борной кислоты, перемешивают 10 с. Оставляют раствор в колбе на 30 мин, время от времени перемешивая.
Раствор переводят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают. Часть раствора фильтруют через двойной фильтр "синяя лента" и отбирают 10 см3 раствора в мерную колбу вместимостью 50 см3. Прибавляют 1 см3 раствора соли Мора и нейтрализуют раствором водного аммиака (1:1) до выпадения гидрооксидов, которые по каплям растворяют в растворе серной кислоты (1:5) и дают избыток 4 см3. Затем добавляют 1 см3 раствора сернокислой меди, 10 см3 раствора тиомочевины и по каплям, непрерывно перемешивая, 4 см3 молибденовокислого аммония.
Ровно через 17 мин разбавляют водой до метки, перемешивают и измеряют оптическую плотность на фотоколориметре с красным или оранжевым светофильтром в кюветах рабочей длиной 50 мм. В качестве холостой пробы или растворов сравнения служит аликвотная часть анализируемого раствора и все реактивы, кроме молибденовокислого аммония.
Все процессы растворения и фильтрования можно вести с большим количеством проб, но после приливания раствора тиомочевины работают не более чем с шестью колбами, т.к. в присутствии раствора сернокислой меди при длительном стоянии растворы становятся мутными.
Параллельно проводят анализ стандартного образца одинаковой навески и близкого по химическому составу и процентному содержанию фосфора к исследуемому.
2.9.4 Расчет результатов анализа
Содержание фосфора (P) в процентах вычисляют по формуле
, (8)
где C - содержание фосфора в стандартном образце, %;
Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора.
2.10 ОПРЕДЕЛЕНИЕ МАРГАНЦА В СТАЛЯХ С СОДЕРЖАНИЕМ ХРОМА МЕНЕЕ 2% И КОБАЛЬТА МЕНЕЕ 0,1%
(по ГОСТ 22536.5)
2.10.1 Сущность метода
Метод основан на окислении двухвалентного марганца в сернофосфорнокислом растворе до семивалентного персульфатом аммония в присутствии азотнокислого серебра как катализатора. Полученную марганцовую кислоту, окрашивающую раствор в характерный фиолетово-красный цвет, оттитровывают раствором тиосульфата натрия. Определению марганца персульфатным методом мешают хром при содержании его более 2% и кобальт при содержании более 0,1%, дающие окраску, на фоне которой трудно установить окончание титрования. При анализе сталей, содержащих более 2% хрома, проводят разделение хрома и марганца окисью цинка.
2.10.2 Реактивы
Кислота серная, пл. 1,84, 1:5.
Кислота фосфорная, пл. 1,7.
Кислота азотная, пл. 1,4.
Смесь кислот: к 700 см3 воды осторожно при непрерывном перемешивании приливают 125 см3 серной кислоты, пл. 1,84, после охлаждения - 175 см3 фосфорной кислоты пл. 1,7.
Серебро азотнокислое, раствор с массовой концентрацией 2 г/дм3.
Аммиак водный, разбавленный 1:1.
Аммоний надсернокислый (персульфат), раствор с массовой концентрацией 200 г/дм3.
Окись цинка, суспензия, 150 г/дм3.
Натрий серноватистокислый, раствор с массовой концентрацией 1,24 г/дм3. Приготовление: 1,24 г натрия серноватистокислого (тиосульфата натрия) растворяют в 1 дм3 свежепрокипяченной охлажденной воды и оставляют на двое суток.
Титр раствора устанавливают по стандартному образцу, содержащему марганец и другие элементы в тех же пределах, что и анализируемые образцы. В конце восстановления марганцевой кислоты титрование следует вести медленно, титрованный раствор прибавлять по каплям и хорошо перемешивать. Титр раствора (T) в граммах в кубическом сантиметре вычисляют по формуле
, (9)
где C - содержание марганца в стандартном образце, %;
mст - навеска стандартного образца, г;
V - объем раствора тиосульфата натрия, израсходованного на титрование марганцевой кислоты, см3.
2.10.3 Проведение анализа
Навеску (стружку) массой 0,2-0,5 г помещают в коническую колбу вместимостью 250 см3, растворяют при нагревании в 30-40 см3 смеси кислот, окисляют тремя-пятью каплями азотной кислоты и кипятят до удаления бурых паров оксидов азота в течение 10-15 мин, разбавляют 60-70 см3 горячей воды, приливают 10 см3 раствора азотнокислого серебра, 20 см3 раствора персульфата аммония, быстро нагревают до кипения и кипятят 0,5-1,0 мин; раствор окрашивается в фиолетово-красный цвет. Колбу снимают с плиты, раствору дают отстояться 2-3 мин для окончания процесса окисления марганца в марганцовую кислоту, затем быстро охлаждают в проточной воде до температуры 15-18 °С. Охлажденный раствор титруют тиосульфатом натрия до полного исчезновения фиолетово-красной окраски.
2.10.4 Расчет результатов анализа
Содержание (Mn) в процентах определяют по формуле
, (10)
где V - объем титрованного раствора тиосульфита натрия, израсходованного на титрование марганцовой кислоты, см3;
T - титр раствора тиосульфата натрия, г/см3;
mисл - навеска исследуемого образца, г.
2.11 ОПРЕДЕЛЕНИЕ МАРГАНЦА В СТАЛЯХ С СОДЕРЖАНИЕМ
ХРОМА БОЛЕЕ 2%
(по ГОСТ 12348)
2.11.1 Сущность метода
См. п.2.10.1.
2.11.2 Реактивы
См. п.2.10.2.
2.11.3 Проведение анализа
Навеску стали массой 0,2-0,5 г помещают в колбу вместимостью 250 см3, приливают 50-70 см3 серной кислоты 1:5 и умеренно нагревают до растворения навески. Приливают по каплям азотную кислоту пл. 1,4 до прекращения вспенивания раствора и достижения избытка ее в количестве двух-трех капель. Окисленный раствор продолжают нагревать до удаления оксидов азота.
Раствор охлаждают, переносят в мерную колбу вместимостью 250 см3. Нейтрализуют избыток кислоты аммиаком до слабощелочной реакции, окраска изменяется на красно-бурую. Затем прибавляют небольшими порциями при непрерывном перемешивании взмученную в воде суспензию цинка до полной коагуляции осадка гидрооксидов и появления на дне колбы белого осадка в виде небольшого кружочка - избыточной окиси цинка. Большого избытка окиси цинка следует избегать. Содержание колбы охлаждают до комнатной температуры, разбавляют до метки водой, перемешивают и дают отстояться.
Раствор фильтруют через сухой фильтр в колбу, сполоснув ее первыми порциями фильтрата. Отбирают аликвотную часть фильтрата, соответствующую навеске стали массой 0,2 г, переносят в коническую колбу вместимостью 250 см3, прибавляют 30 см3 смеси кислот (см. п.2.10.2), разбавляют 60-70 см3 горячей воды и заканчивают определение, как в п.2.10.3.
2.11.4 Расчет результатов
Анализы по п.2.10.4.
2.12 ОПРЕДЕЛЕНИЕ ХРОМА В СТАЛЯХ, НЕ СОДЕРЖАЩИХ ВАНАДИЙ
2.12.1 Сущность метода
Метод основан на окислении трехвалентного хрома персульфатом аммония до шестивалентного в кислой среде в присутствии катализатора - азотнокислого серебра.
Так как ионы хрома окисляются раньше ионов марганца, появление малиновой окраски марганцевой кислоты служит признаком полного окисления хрома. После разрушения марганцовой кислоты хлористым натрием хромовую кислоту титруют раствором соли Мора. В качестве индикатора применяют фенилантраниловую кислоту.
2.12.2 Реактивы
Кислота серная, пл. 1,84. 1:4.
Кислота фосфорная, пл. 1,7.
Кислота азотная, пл. 1,4.
Смесь кислот: к 760 см3 дистиллированной воды осторожно при непрерывном перемешивании приливают 160 см3 серной кислоты пл. 1,84, охлаждают, приливают 80 см3 фосфорной кислоты пл. 1,7 и тщательно перемешивают.
Серебро азотнокислое, раствор с массовой концентрацией 2 г/дм3.
Аммоний надсернокислый (персульфат), раствор с массовой концентрацией 200 г/дм3.
Натрий хлористый, раствор с массовой концентрацией 50 г/дм3.
Кислота фенилантраниловая, раствор с массовой концентрацией 2 г/дм3.
Приготовление 100 см3 раствора: 0,2 г растворяют при нагревании в 100 см3 воды, содержащей 0,2 г углекислого безводного натрия.
Соль закиси железа и аммония двойная сернокислая (соль Мора), раствор с массовой концентрацией эквивалента 0,02 моль/дм3. Приготовление: 8 г соли Мора растворяют в 200-300 см3 воды, прибавляют 50 см3 раствора серной кислоты пл. 1,84, охлаждают и переносят в мерную колбу вместимостью 1 дм3, доводят до метки водой и тщательно перемешивают.
Титр раствора соли Мора (T) в граммах в кубическом сантиметре устанавливают по стандартному образцу стали, соответствующему по химическому составу анализируемому, и вычисляют по формуле
, (11)
где C - содержание хрома в стандартном образце, %;
mст - навеска стандартного образца, г;
V - объем раствора соли Мора, пошедшего на титрование хромовой кислоты, см3.
2.12.3 Проведение анализа
Для определения содержания хрома в стали берут следующие навески:
1,0-0,5 г при содержании хрома 0,2-0,5%;
0,5-0,2 г то же 0,6-2,0%;
0,2-0,1 г -"- 2,1-10%;
0,1 г -"- более 10%.
Взятую навеску помещают в коническую колбу вместимостью 250-750 см3 и растворяют при нагревании в 40 см3 смеси кислот. Раствор окисляют тремя-пятью каплями азотной кислоты пл. 1,4 и кипятят до полного разрушения карбидов и удаления оксидов азота. Приливают 100 см3 горячей воды, 20 см3 раствора азотнокислого серебра, 40 см3 раствора надсернокислого аммония и кипятят до полного окисления хрома (раствор окрашивается в малиновый цвет) и разрушения избытка надсернокислого аммония (появление крупных пузырьков кипения по всему объему раствора); кипячение продолжают в течение 5-10 мин. Для разрушения марганцовой кислоты прибавляют 5 см3 раствора хлористого натрия и кипятят до полного восстановления марганцовой кислоты и коагуляции осадка хлористого серебра; раствор окрашивается в желто-оранжевый цвет. Раствор охлаждают до комнатной температуры, прибавляют 6-10 капель фенилантраниловой кислоты, выдерживают 1 мин и медленно титруют раствором соли Мора до перехода фиолетово-синей окраски в зеленую.
2.12.4 Расчет результатов анализа
Содержание хрома (Cr) в процентах вычисляют по формуле
, (12)
где V - объем раствора соли Мора, израсходованного на титрование, см3;
T - титр раствора соли Мора, г/см3;
mисл - навеска исследуемого образца, г.
2.13 ОПРЕДЕЛЕНИЕ ХРОМА В УГЛЕРОДИСТЫХ СОЛЯХ И СТАЛЯХ, СОДЕРЖАЩИХ ВАНАДИЙ
(по ГОСТ 12350)
2.13.1 Сущность метода
Метод основан на окислении трехвалентного хрома персульфатом аммония до шестивалентного в кислой среде в присутствии катализатора - азотнокислого серебра. Так как ионы хрома окисляются раньше ионов марганца, появление малиновой окраски марганцовой кислоты служит признаком полного окисления хрома. После разрушения марганцовой кислоты хлористым натрием хромовую кислоту восстанавливают раствором соли Мора и избыток соли Мора оттитровывают раствором марганцовокислого калия.
2.13.2 Реактивы
Смесь кислот: к 760 см3 воды осторожно при непрерывном перемешивания приливают 160 см3 серной кислоты пл. 1,84, охлаждают - 80 см3 фосфорной кислоты пл. 1,7 и перемешивают.
Кислота азотная, разбавленная 1:1.
Аммоний надсернокислый (персульфат), раствор с массовой концентрацией 200 г/дм3.
Серебро азотнокислое, раствор с массовой концентрацией 2 г/дм3.
Натрий хлористый, раствор с массовой концентрацией 50 г/дм3.
Соль закиси железа и аммония двойная сернокислая (соль Мора), раствор с массовой концентрацией эквивалента 0,02 моль/дм3: 8 г соли Мора растворяют в 200-300 см3 воды, содержащей 50 см3 серной кислоты пл. 1,84, охлаждают и переносят в мерную колбу вместимостью 1 дм3, доливают до метки, тщательно перемешивают.
Калий марганцовокислый, раствор с массовой концентрацией эквивалента 0,02 моль/дм3: 0,63 г марганцовокислого калия растворяют в 1 дм3 воды в склянке из темного стекла и оставляют отстаиваться в темном месте на 8-10 дней. Отстоявшийся раствор переливают сифоном в другую склянку из темного стекла так, чтобы осадок оксидов марганца остался на дне первой склянки.
Для установления соотношения между растворами соли Мора и марганцовокислого калия в коническую колбу вместимостью 250 см3, содержащую 60 мл серной кислоты 5:95, приливают из бюретки 25 см3 раствора соли Мора и титруют раствором марганцовокислого калия до появления слабо-розовой окраски, устойчивой в течение 1 мин. Соотношение между объемами этих растворов (K) вычисляют по формуле
, (13)
где V2 - объем раствора марганцовокислого калия, израсходованного на титрование, см3;
V1 - объем раствора соли Мора, взятого для титрования, см3.
Титр раствора марганцовокислого калия устанавливают по стандартному образцу стали, близкому по химическому составу и содержанию хрома к исследуемому и прошедшему весь ход анализа.
Титр раствора марганцовокислого калия (T) в граммах в кубическом сантиметре вычисляют по формуле
, (14)
где C - содержание хрома в стандартном образце, %;
mст - навеска стандартного образца, г;
V1 - объем раствора соли Мора, взятого для титрования, см3;
K - соотношение раствора марганцовокислого калия и соли Мора;
V2 - объем раствора марганцовокислого калия, израсходованного на титрование, см3.
2.13.3 Проведение анализа
Навеску стали массой 0,2-1 г (в зависимости от содержания в ней хрома) растворяют в 40 см3 смеси кислот в конической колбе вместимостью 500 см3. Раствор окисляют азотной кислотой пл. 1,4, кипятят до разрушения карбидов и удаления оксидов азота. Добавляют 200 см3 горячей воды, 20 см3 раствора азотнокислого серебра, 40 см3 раствора надсернокислого аммония, нагревают до кипения и кипятят 5-10 мин. После небольшого охлаждения приливают 5 см3 раствора хлористого натрия и продолжают кипятить до восстановления марганцовой кислоты и коагуляции осадка хлористого серебра. Раствор окрашивается в желто-оранжевый цвет. Если окраска марганцовой кислоты не исчезает, то приливают еще 2 см3 раствора хлористого натрия и продолжают кипячение.
После разрушения марганцовой кислоты колбу с содержимым охлаждают до комнатной температуры и, в зависимости от содержания хрома, прибавляют точно отмеренное количество раствора соли Мора до перехода окраски раствора из желто-оранжевой в зеленую и еще дополнительно 5-10 см3. Избыток соли Мора титруют раствором марганцовокислого калия до появления розового оттенка, устойчивого в течение 1 мин, а в присутствии ванадия - в течение 3 мин.
2.13.4 Расчет результатов анализа
Содержание хрома (Cr) в процентах определяют по формуле
, (15)
где T - титр раствора марганцовокислого калия, г/см3, устанавливают по стандартному образцу стали;
K - соотношение раствора марганцовокислого калия и соли Мора;
V1 - объем раствора соли Мора, взятого для титрования, см3;
V2 - объем раствора марганцовокислого калия, израсходованного на титрование, см3;
mисл - навеска исследуемого образца, г.
Примечание. Если в стали содержание хрома превышает 5%, для обратного титрования применяют раствор марганцовокислого калия с массовой концентрацией эквивалента 0,05 моль/дм3 (1,58 г марганцовокислого калия в 1 дм3 воды) и раствор соли Мора с массовой концентрацией эквивалента 0,05 моль/дм3 (20 г соли Мора растворяют в растворе серной кислоты с массовой концентрацией 100 г/дм3).
2.14 ОПРЕДЕЛЕНИЕ ВАНАДИЯ ТИТРИМЕТРИЧЕСКИМ МЕТОДОМ С ФЕНИЛАНТРАНИЛОВОЙ КИСЛОТОЙ
(по ГОСТ 12351)
2.14.1 Сущность метода
Метод основан на окислении четырехвалентного ванадия до пятивалентного марганцовокислым калием, избыток которого восстанавливают щавелевой кислотой или азотистокислым натрием, с последующим титрованием раствором соли Мора в присутствии фенилантраниловой кислоты. Окончание титрования определяют по исчезновению красно-фиолетовой окраски раствора.
2.14.2 Реактивы
Кислота серная пл. 1,84.
Кислота фосфорная пл. 1,7.
Кислота азотная пл. 1,4.
Смесь кислот: к 750 см3 воды осторожно при перемешивании добавляют 150 см3 серной кислоты пл. 1,84, охлаждают и прибавляют 100 см3 фосфорной кислоты пл. 1,7.
Кислота щавелевая, раствор с массовой концентрацией 25 г/дм3.
Кислота фенилантраниловая, раствор с массовой концентрацией 2 г/дм3: 0,2 г растворяют при нагревании в 100 см3 воды, содержащей 0,2 г углекислого безводного натрия.
Соль закиси железа и аммония двойная сернокислая (соль Мора), раствор с массовой концентрацией эквивалента 0,02 моль/дм3: 8 г соли Мора растворяют в 300 см3 воды, содержащей 50 см3 серной кислоты пл. 1,84, охлаждают и переносят в мерную колбу 1 дм3 доливают до метки и перемешивают.
Калий марганцовокислый, раствор с массовой концентрацией 30 г/дм3.
Кислота фенилантраниловая: 0,2 г растворяют при нагревании в 100 см3 воды, содержащей 0,2 г углекислого натрия.
Соль Мора, 0,02 - нормальный раствор: 8 г соли Мора растворяют в 300 см3 воды, прибавляют 50 см3 серной кислоты пл. 1,84 и разбавляют водой до 1 дм3.
Титр раствора устанавливают по стандартному образцу, который проводят через весь ход анализа
, (16)
где C - содержание ванадия в стандартном образце, %;
mст - навеска стандартного образца, г;
V - объем раствора соли Мора, израсходованного на титрование, см3.
2.14.3 Проведение анализа
Навеску стали массой 1 г помещают в коническую колбу вместимостью 250 см3 и растворяют в 70 см3 смеси кислот: прибавляют 1-2 см3 азотной кислоты пл. 1,4 и кипятят до разрушения карбидов. Раствор упаривают до выделения густых паров сернистого ангидрида, охлаждают, разбавляют 100 см3 воды и нагревают до растворения солей. Раствор охлаждают и прибавляют по каплям раствор марганцовокислого калия до получения устойчивой малиновой окраски, которая не исчезает в течение 2-3 мин. Избыток марганцовокислого калия восстанавливают щавелевой кислотой, прибавляя по каплям до полного обесцвечивания раствора. К раствору, имеющему зеленую окраску без розового оттенка, прибавляют 5 капель фенилантраниловой кислоты и 10 см3 серной кислоты (1:1) для сталей, содержащих менее 0,3% ванадия. В присутствии ванадия раствор окрашивается в темно-вишневый цвет. Спустя 2-3 мин полученный раствор титруют раствором соли Мора до перехода темно-вишневой окраски в желто-зеленую, а при содержании хрома более 1% - в изумрудно-зеленую.
2.14.4 Расчет результатов анализа
Содержание ванадия (V) в процентах вычисляют по формуле
, (17)
где T - титр раствора соли Мора, г/см3;
V - объем титрованного раствора соли Мора, израсходованного на титрование, см3;
mисл - навеска исследуемого образца, г.
2.15 ОПРЕДЕЛЕНИЕ МАРГАНЦА, ХРОМА И ВАНАДИЯ ИЗ ОДНОЙ НАВЕСКИ
2.15.1 Сущность метода
См. пп.2.10.1; 2.13.1 и 2.14.1.
2.15.2 Реактивы
См. пп.2.10.2; 2.13.2 и 2.14.2.
2.15.3 Проведение анализа
Навеску стали массой 1 г растворяют в 40 см3 смеси кислот (см. п.2.13.2) в конической колбе вместимостью 250-750 см3, прибавляют 1-2 см3 азотной кислоты пл. 1,4 и кипятят до разрушения карбидов и удаления оксидов азота. Добавляют 100 см3 горячей воды, 20 см3 раствора азотнокислого серебра и 40 см3 персульфата аммония. Раствор быстро охлаждают в проточной воде, добавляют 2 см3 фосфорной кислоты пл. 1,7 и оттитровывают марганцовую кислоту титрованным раствором тиосульфата натрия. Для разложения избытка тиосульфата натрия раствор кипятят и повторяют окисление персульфатом аммония с добавлением раствора азотнокислого серебра. Образовавшуюся марганцовую кислоту разрушают добавлением 5 см3 раствора хлористого натрия при нагревании. После охлаждения оттитровывают хром и ванадий титрованным раствором соли Мора в присутствии индикатора - раствора фенилантраниловой кислоты (V1 - объем раствора соли Мора (см3), затраченного на титрование хрома и ванадия).
В оттитрованном растворе окисляют ванадий раствором перманганата калия до пятивалентного состояния. Избыток перманганата калия разрушают раствором щавелевой кислоты, которую прибавляют по каплям до исчезновения розовой окраски. Добавляют 2-3 капли раствора фенилантраниловой кислоты и титруют раствором соли Мора, с массовой концентрацией эквивалента 0,02 моль/дм3 (V2) до перехода темно-вишневой окраски в желто-зеленую (V2 - объем раствора соли Мора, затраченного на титрование ванадия).
Объем раствора соли Мора, затраченного на титрование хрома, определяют как разность V1 – V2.
2.15.4 Расчет результатов анализа
Для марганца | - см. п.2.10.4; |
для хрома | - см. п.2.12.4; |
для ванадия | - см. п.2.14.4. |
2.16 ОПРЕДЕЛЕНИЕ МОЛИБДЕНА ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
(по ГОСТ 12354)
2.16.1 Сущность метода
Метод основан на образовании комплексного соединения пятивалентного молибдена с роданистым аммонием. Молибден восстанавливают до пятивалентного тиомочевиной в сернокислом растворе в присутствии ионов меди. Определению молибдена мешает вольфрам, который образует зеленовато-желтый роданид вольфрама.
2.16.2 Реактивы
Кислота серная, пл. 1,84, 1:4.
Кислота азотная, пл. 1,4.
Кислота соляная, пл. 1,19.
Медь сернокислая, раствор с массовой концентрацией 10 г/дм3.
Тиомочевина, раствор с массовой концентрацией 100 г/дм3.
Аммоний роданистый, с массовой концентрацией 250 г/дм3, свежеприготовленный.
Смесь кислот; к 750 см3 воды, осторожно помешивая, добавляют 200 см3 серной кислоты пл. 1,84, охлаждают и добавляют 50 см3 соляной кислоты пл. 1,19.
2.16.3 Проведение анализа
Навеску стали массой 0,25 г растворяют при умеренном нагревании в 40-50 см3 серной кислоты 1:4 в стакане вместимостью 200 см3, окисляют одной-тремя каплями азотной кислоты, выпаривают до паров серной кислоты и охлаждают. Затем приливают 10-15 см3 воды и снова нагревают до паров серной кислоты. После охлаждения приливают 15-20 см3 воды, омывая ею стенки стакана, и, накрыв стакан часовым стеклом, кипятят несколько минут для растворения солей. Охлажденный раствор переводят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают. Отбирают аликвотную часть раствора 2-5 см3 - в мерную колбу вместимостью 100 см3, прибавляют 40 см3 смеси кислот, 1 см3 раствора сернокислой меди, 10 см3 раствора тиомочевины и через 5 мин - 4 см3 раствора роданистого аммония. После прибавления каждого реактива раствор тщательно перемешивают. Через 30 мин окрашенный раствор фотоколориметрируют с синим светофильтром в кювете рабочей длиной 30 мм.
В качестве раствора сравнения применяют вторую аликвотную часть исследуемого раствора, в которую добавляют все реактивы, кроме роданистого аммония. Параллельно проводят анализ стандартного образца, близкого по химическому составу к исследуемому.
2.16.4 Расчет результатов анализа
Содержание молибдена (Mo) в процентах вычисляют по формуле
, (18)
где Mo - содержание молибдена в исследуемом образце, %;
Dисл - оптическая плотность исследуемого раствора;
C - содержание молибдена в стандартом образце, %;
mст - навеска стандартного образца, соответствующая аликвотной части, г;
Dст - оптическая плотность стандартного раствора;
mисл - навеска исследуемого образца, соответствующая аликвотной части, г.
Расчет можно производить также по градуировочному графику.
2.16.5 Построение градуировочного графика
Стандартный образец массой 1 г с содержанием молибдена 0,5% растворяют в 50 см3 серной кислоты (1:4), дважды упаривают до паров серной кислоты, охлаждают, приливают 20 см3 воды для растворения солей и переводят в мерную колбу вместимостью 250 см3. В десять мерных колбочек вместимостью по 100 см3 вливают из микробюретки аликвотные части стандартного раствора 0,5; 1,0; 1,5+ 5,0 см3. Далее анализ проводят по п.2.16.3.
Примечание: 1. При наличии в стали вольфрама для растворения солей прибавляют 10 см3 раствора лимоннокислого аммония двузамещенного с массовой концентрацией 30 г/дм3 и строят градуировочный график с учетом присутствия вольфрама.
2. Содержание вольфрама до 1 мг в колориметрируемом объеме не мешает определению.
3. До полного восстановления железа, молибдена и развития окраски раствор выдерживают при анализе сплавов на никелевой основе 20-30 мин, на железной основе - 30-60 мин.
4. В 50 см3 колориметрируемого раствора не должно содержаться более 0,06 мг молибдена; при более высоких его концентрациях наблюдается несоответствие закону Ламберта-Бера.
5. При использовании кювет рабочей длины 30 мм оптическая плотность не должна превышать значения 0,23. Наиболее сходимые результаты получаются при оптической плотности 0,07-0,18.
2.17 ОПРЕДЕЛЕНИЕ ТИТАНА ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ С ДИАНТИПЕРИЛМЕТАНОМ
(по ГОСТ 12356)
2.17.1 Сущность метода
Четырехвалентный титан образует с диантиперилметаном комплексное соединение, окрашенное в желто-оранжевый цвет. Влияние трехвалентного железа и пятивалентного ванадия устраняют прибавлением аскорбиновой кислоты.
2.17.2 Реактивы
Кислота соляная, пл. 1,19, 1:1.
Кислота азотная, пл. 1,4.
Смесь кислот: 3 части соляной кислоты пл. 1,19, 1 часть азотной кислоты пл. 1,4.
Кислота серная пл. 1,84; 1:1, 1:4.
Кислота аскорбиновая, раствор с массовой концентрацией 100 г/дм3.
Диантиперилметан, раствор с массовой концентрацией 10 г/дм3, свежеприготовленный. Приготовление: 10,0 г диантиперилметана растворяют в 30-40 см3 воды, содержащей 15 см3 серной кислоты пл. 1,84. Раствор фильтруют в мерную колбу вместимостью 1 дм3, разбавляют водой до метки и перемешивают.
Кислота фосфорная, пл. 1,7.
2.17.3 Проведение анализа
Навески стали массой 0,5 г при содержании титана 0,1-1,0% помещают в стакан вместимостью 200-250 см3 и растворяют в 25 см3 серной кислоты 1:4. Если проба не растворяется, берут другую навеску и растворяют в 15 см3 смеси кислот. Прибавляют 15 см3 серной кислоты (1:1) и выпаривают раствор до паров серной кислоты.
Раствор переносят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают. Часть раствора отфильтровывают через два сухих фильтра "белая лента", отбрасывая первые порции фильтрата.
Аликвотную часть раствора, равную 5 см3 при содержании титана 0,5% и 10 см3 при 0,2%, помещают в мерную колбу вместимостью 100 см3, добавляют 5 см3 раствора аскорбиновой кислоты, перемешивают и оставляют на 5-7 мин до полного восстановления железа и ванадия; прибавляют 10 см3 соляной кислоты (1:1) для разрушения окрашенного соединения титана с аскорбиновой кислотой и 25 см3 раствора диантиперилметана, доливают до метки водой и перемешивают. Оптическую плотность полученного раствора измеряют через 1 ч на фотоколориметре с сине-зеленым светофильтром в кювете рабочей длиной 50 мм.
Содержание титана находят методом сравнения со стандартным образцом стали, близким по содержанию титана к исследуемому, прошедшим весь ход анализа, или по калибровочному графику. В качестве холостой пробы используют исследуемый раствор с добавлением всех реактивов, кроме диантиперилметана.
2.17.4 Расчет результатов анализа
Содержание титана (Ti) в процентах вычисляют по формуле
, (19)
где C - содержание титана в стандартном образце, %;
Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора.
2.17.5 Построение градуировочного графика
Стандартный образец массой 1 г, близкий по содержанию титана к исследуемым пробам, растворяют, как описано в п.2.17.3. Из микробюретки берут аликвотные части стандартного раствора, равные 5,0; 6,0... 15 см3, переводят в мерные колбы вместимостью по 100 см3, прибавляют 5 см3 раствора аскорбиновой кислоты, 10 см3 соляной кислоты (1:1), 25 см3 раствора диантиперилметана. Доливают водой до метки, тщательно перемешивают и измеряют оптическую плотность в кюветах рабочей длиной 50 мм с сине-зеленым фильтром.
Примечание. Оставшийся раствор после отбора аликвотной части для определения титана может быть использован для определения марганца, хрома, никеля методами, описанными в пп.2.11, 2.12, 2.13 и 2.19.
2.18 ОПРЕДЕЛЕНИЕ НИКЕЛЯ ГРАВИМЕТРИЧЕСКИМ МЕТОДОМ ПРИ ЕГО СОДЕРЖАНИИ В СТАЛИ 0,6-30%
(по ГОСТ 12352)
2.18.1 Сущность метода
Метод основан на осаждении никеля диметилглиоксимом в слабощелочном растворе в присутствии комплексообразователей винной или лимонной кислоты, сегнетовой соли или кали-натрия виннокислого с последующим взвешиванием осадка в воде диметилглиоксимата никеля.
2.18.2 Реактивы
Кислота соляная, пл. 1,19; 1:1, 1:100.
Кислота азотная, пл. 1,4.
Аммиак водный.
Кислота винная, раствор в массовой концентрацией 25 г/дм3.
Диметилглиоксим, спиртовой раствор с массовой концентрацией 10 г/дм3. Приготовление: 10 г диметилглиоксима растворяют в 750 м3 спирта, приливают 250 см3 горячей воды и перемешивают (или 10 г диметилглиоксима растворяют в гидроксиде натрия с массовой концентрацией 50 г/дм3 или аммиака водного, разбавленного 1:1).
Метиловый оранжевый, раствор с массовой концентрацией 1 г/дм3.
2.18.3 Проведение анализа
Для определения никеля в стали берут навески:
1 г при содержании никеля до 1,5%;
0,5 г то же 1,5-5,0%;
0,2 г при содержании никеля 5,1-18%;
0,1 г то же более 18%.
Навеску стали (стружку) помещают в коническую колбу вместимостью 750 см3 и растворяют при умеренном нагревании в 30 см3 соляной кислоты (1:1). После растворения стружки осторожно по каплям прибавляют азотную кислоту пл. 1,4 до прекращения вспенивания раствора и выделения оксида азота. Если в стали присутствуют вольфрам и ниобий, их выделяют гидролизом, осадок отфильтровывают на фильтр "синяя лента", промывают водой и отбрасывают.
При содержании кремния в стали более 1% раствор выпаривают досуха. Сухой остаток растворяют в 30 см3 соляной кислоты пл. 1,19 и 100 см3 воды, осадок отфильтровывают на фильтре "белая лента", промывают водой и отбрасывают.
Если сталь содержит не более 1% вольфрама, ниобия и кремния, после выделения бурых паров оксида азота, раствор разбавляют водой до 300 см3, прибавляют 20-30 см3 раствора винной кислоты и осторожно нейтрализуют водным раствором аммиака до слабого запаха. В нагретом до температуры 60 °С растворе осаждают никель диметилглиоксимом, прибавляя его в количестве 30-40 см3. Колбы с осадком ставят на 30-40 мин в теплое место. Осадок отфильтровывают на фильтр средней плотности, промывают несколько раз теплой водой. Осадок на фильтре растворяют горячей соляной кислотой (1:1) и собирают в колбу, в которой производилось осаждение; фильтр хорошо промывают горячей водой. Раствор разбавляют водой до 200-300 см3 и повторно прибавляют 20 см3 раствора винной кислоты, нейтрализуют аммиаком по метиловому оранжевому до слабо щелочной реакции, приливают 30 см3 раствора диметилглиоксима и аммиак до его явного запаха. Тщательно перемешивают и дают отстояться в течение 15-20 мин.
Осадок отфильтровывают, промывают 8-10 раз теплой водой и переносят во взвешенный тигель; просушивают при температуре 110-120 °C, осторожно озоляют и прокаливают при температуре 800-825 °С до постоянной массы.
2.18.4 Расчет результатов анализа
Содержание никеля (Ni) в процентах вычисляют по формуле
, (20)
где A - масса осадка закиси никеля, г;
0,7858 - коэффициент пересчета закиси никеля на никель;
mисл - навеска исследуемого образца, г.
2.19 ОПРЕДЕЛЕНИЕ НИКЕЛЯ ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
(по ГОСТ 12352)
2.19.1 Сущность метода
Метод основан на образовании растворимого соединения никеля с диметилглиоксимом в щелочной среде в присутствия окислителя, персульфата аммония. Вредное влияние железа, хрома, кобальта и меди устраняется прибавлением к исследуемому раствору сегнетовой соли или винной кислоты.
Присутствие более 4-5% марганца мешает определению, образуя муть. Можно отделить никель от мешающих элементов осаждением его диметилглиоксимом с последующим растворением осадка в соляной кислоте и определением никеля фотоколориметрическим методом.
2.19.2 Реактивы
Кислота соляная, 1:1.
Кислота азотная, пл. 1,4; 1:3.
Кислота серная, 1:4.
Сегнетовая соль, раствор с массовой концентрацией 200 г/дм3.
Натрия гидроксид, раствор с массовой концентрацией 100 г/дм3.
Персульфат аммония, раствор с массовой концентрацией 100 г/дм3.
Диметилглиоксим, раствор с массовой концентрацией 10 г/дм3, в растворе гидроксида натрия с массовой концентрацией 50 г/дм3.
2.19.3 Проведение анализа
Навеску стали массой 0,1-0,2 г растворяют при нагревании в 10-25 см3 азотной кислоты (1:3) и кипятят до удаления оксида азота. Стали, плохо растворимые в азотной кислоте, растворяют в 10-20 см3 соляной кислоты (1:1) с последующим окислением азотной кислотой пл. 1,4. Сталь с большим содержанием хрома (10% и более) растворяют в серной кислоте 1:4, после полного растворения навески окисляют 5-10 каплями азотной кислоты. Полученный раствор переводят в мерную колбу вместимостью 100 см3 и разбавляют водой до метки.
Для определения никеля в мерную колбу вместимостью 100 см3 отбирают аликвотную часть раствора, содержащую 0,01-0,1-0,2 мг никеля, прибавляют 10 см3 раствора сегнетовой соли, 10 см3 раствора гидроксида натрия, 10 см3 раствора персульфата аммония и 10 см3 раствора диметилглиоксима, каждый раз перемешивая раствор, выдерживают его 5 мин до полного развития окраски и доводят до метки водой. Оптическую плотность измеряют на фотоколориметре с синим светофильтром в кюветах рабочей длиной 50 мм или 5 мм при содержании никеля соответственно от 0,1 до 10% или от 10 до 60%.
Раствором сравнения служит соответствующая аликвотная часть исследуемого раствора, содержащая все реактивы, кроме диметилглиоксима.
Содержание никеля рассчитывают по стандартному образцу, близкому по содержанию никеля к исследуемому.
2.19.4 Расчет результатов анализа
Содержание никеля (Ni) в процентах определяют по формуле
, (21)
где Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора;
C - содержание никеля в стандартном образце, %;
mст - навеска стандартного образца, соответствующая аликвотной части, г;
mисл - навеска исследуемого раствора, г.
Содержание никеля можно рассчитывать также по градуировочному графику.
2.19.5 Построение градуировочного графика
Для построения графика берут навеску массой 0,2 г стандартного образца с содержанием никеля 99,9%, растворяют в 20 см3 азотной кислоты 1:1, охлаждают, переливают в мерную колбу вместимостью 0,5 дм3, доливают до метки водой и тщательно перемешивают. Отбирают в две мерные колбы вместимостью по 1 дм3 аликвотные части: 25 см3 с титром 0,00001 г никеля в 1 см3 для построения градуировочного графика при определении никеля от 0,1 до 15% и 250 см3 с титром 0,0001 г никеля в 1 см3 при определении никеля от 15 до 60%.
В мерные колбочки вместимостью по 100 см3 приливают от 0,5 до 8 см3 стандартного раствора с интервалами в 0,5 см3. В каждую колбочку при перемешивании приливают реактивы, как указано в п.2.19.3. Через 5 мин растворы разбавляют до метки водой, перемешивают и колориметрируют с синим светофильтром.
2.20 ОПРЕДЕЛЕНИЕ ВОЛЬФРАМА ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
(по ГОСТ 12349)
2.20.1 Сущность метода
Метод основан на образовании пятивалентного соединения вольфрама с роданидом калия или аммония в кислой среде, окрашенного в зеленовато-желтый цвет. Катионы железа, хрома и никеля, мешающие определению, отделяют едким натром.
2.20.2 Реактивы
Кислота соляная, пл. 1,19; 1:1.
Кислота азотная, пл. 1,4.
Аммоний роданистый, раствор с массовой концентрацией 250 г/дм3.
Натрия гидроксид, раствор с массовой концентрацией 200 г/дм3.
Титан треххлористый, раствор с массовой концентрацией 50 г/дм3 в растворе соляной кислоты, разбавленной 1:1. Приготовление: 5 см3 треххлористого титана разбавляют до 100 см3 соляной кислотой 1:1 или 1 г металлического титана растворяют в 60 см3 соляной кислоты (1:1), раствор переливают в мерную колбу вместимостью 100 см3 и разбавляют водой до метки.
2.20.3 Проведение анализа
Навеску стали массой 0,25 г помещают в стакан вместимостью 300 см3 и растворяют в 30 см3 соляной кислоты 1:1, окисляют азотной кислотой. Прибавляют 3 см3 серной кислоты пл. 1,84, выпаривают до появления белых паров серной кислоты, охлаждают и прибавляют 50 см3 дистиллированной воды. Растворение солей ведут при нагревании, затем нейтрализуют раствором гидроксида натрия до появления осадка, который растворяют в нескольких каплях серной кислоты 1:4, раствор переливают в мерную колбу вместимостью 200 см3, содержащую 30 см3 раствора гидроксида натрия, осторожно перемешивают. Гидрооксиды железа, никеля, хрома и титана выпадают в осадок. Раствор охлаждают и разбавляют водой до метки. Отфильтровывают часть раствора через сухой фильтр, первые порции фильтрата отбрасывают.
Затем в мерную колбу вместимостью 50 см3 отбирают пипеткой 5-20 см3 фильтрата в зависимости от предполагаемого содержания вольфрама, прибавляют 20 см3 соляной кислоты (1:1), охлаждают, прибавляют 2-3 см3 раствора роданида аммония, выдерживают 5 мин и прибавляют пять капель раствора треххлористого титана до исчезновения окраски комплексного соединения молибдена с роданидом и избыток его пять капель до появления зеленовато-желтой окраски. Доливают до метки соляной кислотой (1:1), перемешивают и через 30 мин измеряют оптическую плотность раствора в кюветах рабочей длиной 50 мм с синим светофильтром.
Расчет содержания вольфрама ведут по стандартному образцу, близкому по содержанию вольфрама к исследуемому образцу.
2.20.4 Расчет результатов анализа
Содержание вольфрама (W) в процентах вычисляют по формуле
, (22)
где C - содержание вольфрама в стандартном образце, %;
Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора.
Содержание вольфрама можно рассчитывать также по градуировочному графику.
2.20.5 Построение калибровочного графика
Для построения графика 0,1360 г вольфрамовой кислоты растворяют в 50 см3 раствора гидроксида натрия, разбавляют дистиллированной водой до 1 дм3 и перемешивают. В мерные колбочки вместимостью по 50 см3 отбирают из микробюретки стандартный раствор вольфрама от 0,2 до 3,0 см3 с интервалом 0,2 см3. Прибавляют по 1 см3 раствора гидроксида натрия и далее все реактивы, указанные в п.2.19.3. По показаниям шкалы фотоколориметра строят калибровочный график.
2.21 ОПРЕДЕЛЕНИЕ НИОБИЯ ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ С ЦИАНФОРМАЗАНОМ-2
2.21.1 Сущность метода
Ниобий образует с цианформазаном окрашенное в синий цвет соединение. Ниобий можно определять в присутствии 12-15-кратных количеств вольфрама, 30-кратных тантала, 40-50-кратных титана и железа. Алюминий, никель, марганец не мешают определению, даже если их количество превышает содержание ниобия более чем в 600 раз.
2.21.2 Реактивы
Кислота соляная, пл. 1,19; разбавленная 1:10, 1:25.
Смесь кислот; 3 части соляной кислоты пл. 1,19 и 1 часть азотной кислоты пл. 1,4.
Кислота азотная, пл. 1,4.
Кислота фениларсоновая, раствор с массовой концентрацией 25 г/дм3.
Калий пиросернокислый.
Кислота винная, раствор с массовой концентрацией 200 г/дм3.
Трилон Б, раствор с массовой концентрацией эквивалента 0,02 моль/дм3.
Цианформазан-2, раствор с массовой концентрацией 3 г/дм3.
2.21.3 Проведение анализа
Навеску стали массой 0,3 г растворяют в 30 см3 смеси кислот. После полного растворения навески раствор выпаривают досуха и слегка высушивают осадок; добавляют 15 см3 соляной кислоты пл. 1,19 и растворяют осадок при нагревании. Раствор с осадком разбавляют горячей водой температурой 75 °С до 200 см3 и нагревают до кипения. Приливают 40 см3 раствора фениларсоновой кислоты для доосаждения ниобия и оставляют на ночь. Выделившийся осадок отфильтровывают на двойной плотный фильтр с бумажной массой и промывают горячей разбавленной 1:25 соляной кислотой. Осадок помещают в тигель, озоляют, сплавляют с 2-3 г пиросернокислого калия и выдерживают 10 мин при температуре 700 °С. Плав выщелачивают в 15-20 см3 раствора винной кислоты.
Раствор переводят в мерную колбу вместимостью 100 см3, разбавляют водой до метки и перемешивают. Если при выщелачивании в винной кислоте раствор получился мутный, его отфильтровывают.
Для анализа 2 см3 раствора вливают в мерную колбу вместимостью 100 см3, прибавляет 10 см3 раствора соляной кислоты (1:10), 1 см3 раствора трилона Б для связывания мешающих определению компонентов, 2 см3 водного раствора цианформазана-2 и добавляют до метки раствором соляной кислоты (1:10) и перемешивают.
Раствор нагревают на водяной бане в течение 30 мин при температуре 60 °С, охлаждают до комнатной температуры и измеряют оптическую плотность на фотоколориметре с красным светофильтром в кюветах рабочей длиной 30 мм.
Расчет результатов анализа ведут по стандартному образцу, близкому по химическому составу к исследуемому, или по градуировочному графику.
2.21.4 Расчет результатов анализа
Содержание ниобия (Nb) в процентах вычисляют по формуле
, (23)
где C - содержание ниобия в стандартном образце, %;
Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора.
2.21.5 Построение градуировочного графика
Для построения градуировочного графика 0,3 г стандартного образца растворяют в 30 см3 смеси кислот. Для построения графика отбирают аликвотные части 0,5; 1,0... 5,0 см3 и проводят через весь ход анализа (см. п.2.21.3).
2.22 ОПРЕДЕЛЕНИЕ МЕДИ ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
(ГОСТ 22536.8)
2.22.1 Сущность метода
Метод основан на реакции взаимодействия меди с диэтилдитиокарбаматом натрия с образованием при показателе рН, равном 8,5, комплексного соединения диэтилдитиокарбамата меди желто-коричневого цвета. Для стабилизации комплекса применяют желатину.
2.22.2 Реактивы
Кислота азотная, 1:3.
Кислота соляная, пл. 1,19.
Натрий пирофосфорнокислый, раствор с массовой концентрацией 100 г/дм3.
Аммиак, водный раствор.
Желатина, раствор с массовой концентрацией 5 г/дм3.
Диэтилдитиокарбамат натрия, раствор с массовой концентрацией 5 г/дм3.
2.22.3 Проведение анализа
Навеску стали массой 0,25 г растворяют при нагревании в 20 см3 азотной кислоты 1:3. После растворения прибавляют 5 см3 соляной кислоты пл. 1,19 и выпаривают досуха. К сухому осадку прибавляют 15 см3 соляной кислоты пл. 1,19, 10 см3 воды и нагревают до полного растворения осадка. Раствор охлаждают, переливают в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают. Затем 10 см3 раствора переносят в другую мерную колбу вместимостью 100 см3, приливают 20 см3 воды, 20 см3 раствора пирофосфата натрия, 10 см3 водного раствора аммиака, 10 см3 раствора желатины, 10 см3 раствора диэтилдитиокарбамата натрия, доливают до метки водой и перемешивают. Оптическую плотность измеряют с синим светофильтром в кювете рабочей длиной 30 мм.
Раствором сравнения служит испытуемый раствор с добавлением всех реактивов, кроме диэтилдитиокарбамата натрия. Содержание меди определяют по стандартному образцу.
2.22.4 Расчет результатов анализа
Содержание меди (Cu) в процентах вычисляют по формуле
, (24)
где C - содержание меди в стандартном образце, %;
Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора.
3 ФАЗОВЫЙ АНАЛИЗ ЭНЕРГЕТИЧЕСКИХ СТАЛЕЙ
3.1 ОБЩИЕ ПОЛОЖЕНИЯ
Методы физико-химического фазового анализа широко применяют при решении конкретных проблем связи структуры и фазового состава со свойствами сталей.
Основной частью физико-химического фазового анализа является электрохимическое или химическое выделение карбидных и интерметаллидных фаз и последующий их химический и рентгеноструктурный анализ.
Эти методы наиболее доступны и позволяют получить наиболее объективную и точную информацию о природе, составе и количестве исследуемых фаз, хотя выбор электролитов и режимов анодного растворения для конкретных случаев комбинаций карбидных и интерметаллидных фаз в сталях, как правило, проводят эмпирически.
Для изучения фазового состава энергетических сталей применяют следующие методы, основное различие которых состоит в способе выделения фаз и выражения результатов анализа:
электролитическое растворение специально изготовленного образца;
выделение карбидов методом химического растворения стружки стали, снятой с поверхности исследуемой детали;
химическое растворение стали с участка поверхности детали без нарушения ее целостности. Этот метод позволяет установить распределение элементов между карбидной фазой и твердым раствором. Расчет ведется по соотношению содержания элементов в основной и карбидной фазах. Метод из-за малой поверхности растворения может служить лишь как полуколичественный метод определения карбидов.
3.2 ПОДГОТОВКА ОБРАЗЦОВ ДЛЯ ФАЗОВОГО AHAЛИЗA
Для фазового анализа применяют образцы стали цилиндрической формы диаметров 8-10 мм, длиной 60-80 мм с отверстием диаметром 2 мм на расстоянии 4-5 мм от конца образца. Образцы можно изготовить в виде брусков таких же размеров с закругленными гранями.
Поверхность образца должна быть чистой (без окалины), без грубых рисок.
3.3 ИЗГОТОВЛЕНИЕ КОЛЛОДИЕВОГО МЕШОЧКА
Мешочки для водных электролитов изготовляют из коллодия, которым заполняют специальную круглодонную пробирку диаметром 40-45 мм и высотой 100-120 мм, избыток коллодия выливают. Вращением пробирки достигают равномерного распределения коллодия по ее стенкам. Коллодиевую пленку подсушивают вращением пробирки между ладонями. Мешочек осторожно отделяют от стенок пробирки и быстро прикрепляют к стеклянному кольцу. Диаметр кольца должен соответствовать диаметру пробирки. Готовый мешочек заполняют водой для проверки его целости и хранят под водой.
3.4 ЭЛЕКТРОЛИТИЧЕСКИЙ МЕТОД ФАЗОВОГО АНАЛИЗА С ИЗГОТОВЛЕНИЕМ СПЕЦИАЛЬНОГО ОБРАЗЦА
Фазовый анализ основан на электролитическом, избирательном выделении фаз с последующим изучением анодного осадка.
Электролитическое изолирование фаз происходит при растворении образца стали, через который пропускают постоянный ток, в подобранном для данной стали электролите. Катодом служит пластина цилиндрической формы из нержавеющей стали или меди, анодом - образец стали. На аноде происходит избирательное растворение одних фаз и выделение на поверхности образца в виде черного или серого осадка других фаз.
Исследуемый образец помещают в полупроницаемую мембрану - коллодиевый мешочек, в который собирают нерастворимый осадок, а ионы растворенной стали сквозь поры мембраны проникают в электролит.
Электролиз ведут в фарфоровом стакане или кружке вместимостью 1 дм3, заполненной выбранным электролитом. По стенке электролитического сосуда расположен катод. В сосуд опускают мешочек на стеклянном каркасе. Мешочек заполняют электролитом и опускают в него образец, прикрепленный проволочкой к медной пластине, предварительно взвешенный на аналитических весах. Перед началом электролиза электролит, если это требуется, охлаждают до определенной температуры; охлаждение производят смесью снега (льда) с различными солями (хлористым калием, хлористым натрием и другими), располагая охлаждающую смесь вокруг электролитического сосуда. Образец во время электролиза располагают в центре коллодиевого мешочка. Чтобы не происходило бурного растворения образца на границе воздух - электролит, на концы образца надевают резиновые трубочки, закрывающие нижний конец образца и участок на границе воздух - электролит.
Электролиз ведут в течение 2-3 ч при плотности тока 0,02-0,05 А/см2 площади поверхности образца и 10-15 мин при плотности тока 0,3-0,5 А/см2. В зависимости от площади поверхности образца продолжительность электролиза может быть уменьшена, количество растворенной для анализа стали должно быть не менее 1,5-2,0 г для перлитных сталей и 1,0-1,5 г для аустенитных сталей.
Установка для электролитического растворения состоит из селенового выпрямителя ВСА-6А, амперметра и реостата для регулирования силы тока. Амперметр и реостат выбирают в соответствии с требуемой для электролиза силой тока. Схема установки представлена на рисунках 3 и 4.
1 - фарфоровый стакан; 2 - катод из нержавеющей стали; 3 - коллодиевый мешочек;
4 - исследуемый образец; 5 - стеклянный каркас для коллодиевого мешочка; 6 - амперметр;
7 - реостат; 8 - селеновый выпрямитель
Рисунок 3 - Схема установки для электролитического растворения параллельно соединенных образцов для фазового анализа
Рисунок 4 - Схема установки для электролитического растворения последовательно соединенных образцов для фазового анализа (пояснения к рисунку см. рисунок 3).
При последовательном соединении на все образцы подают ток одинаковой силы, поэтому площадь поверхности образцов должна быть одинаковой. При параллельном соединении каждая электролитическая ячейка имеет свой амперметр и peocтат, что позволяет регулировать силу тока индивидуально для каждого образца. Осадки с образцов снимают механическим путем стеклянной палочкой с резиновым наконечником или ультразвуком.
Примечания: 1. Установка для электролитического выделения анодного осадка, метода подготовки образцов для анализа и снятия осадка - одинаковые для всех марок стали.
2. Электролитическая установка должна быть снабжена вентиляцией.
3.5 ФАЗОВЫЙ АНАЛИЗ ПЕРЛИТНЫХ СТАЛЕЙ
(12MX, 15XM, 12Х1МФ, 15Х1М1Ф, 20ХМФЛ)
3.5.1 Условия электролиза
В перлитных сталях в процессе термообработки и эксплуатационного старения образуются фазы типа Ме3C, Mе7C3, Ме23С6, МеС, Ме2С. Для изолирования фаз из хромомолибденовых и хромомолибденованадиевых сталей применяют электролит следующего состава: 75 г/дм3 хлористого калия, 5 г/дм3 лимонной кислоты; температура электролита - 10-16 °С; плотность тока 0,02-0,05 А/см2, продолжительность электролиза 2-3 ч.
В этом электролите при соблюдении указанных условий электролиза изолируются все вышеперечисленные фазы. Для изолирования мелкодисперсных фаз и сохранения их в процессе электролиза в осадке необходимо понижать температуру электролита до 0 °С.
Для отделения изолированных фаз одну от другой применяют химическую обработку выделенного осадка соответствующими реагентами в зависимости от того, какие фазы надо сохранить в осадке, а какие - растворить.
3.5.2 Реактивы
Электролит: 75 г/дм3 хлористого калия, 5 г/дм3 лимонной кислоты.
Калий пиросернокислый.
Кислота серная пл. 1,84; 1:4, 1:1.
Кислота фосфорная, пл. 1,7.
Кислота азотная, пл. 1,4.
Смесь кислот; к 800 см3 воды осторожно при перемешивании прибавляют 120 см3 серной кислоты пл. 1,84 и после охлаждения 80 см3 фосфорной кислоты пл. 1,7.
Аммиак, раствор с массовой концентрацией 250 г/дм3.
Серебро азотнокислое, раствор с массовой концентрацией 2 г/дм3.
Аммоний надсернокислый, раствор с массовой концентрацией 200 г/дм3.
Натрий хлористый, раствор с массовой концентрацией 50 г/дм3.
Двойная сернокислая соль закиси железа и аммония (соль Мора), раствор с массовой концентрацией эквивалента 0,02 моль/дм3, приготовление см. п.2.12.2.
Калий марганцовокислый, стандартный раствор с массовой концентрацией эквивалента 0,02 моль/дм3.
Кислота фенилантраниловая, раствор с массовой концентрацией 2 г/дм3; приготовление см. п.2.14.2.
Кислота щавелевая, раствор с массовой концентрацией 20 г/дм3.
Медь сернокислая, раствор с массовой концентрацией 10 г/дм3.
Тиомочевина, раствор с массовой концентрацией 100 г/дм3, свежеприготовленный.
Аммоний роданистый, раствор с массовой концентрацией 250 г/дм3, свежеприготовленный.
Натрий серноватистокислый (тиосульфат натрия), раствор с массовой концентрацией 1,24 г/дм3; приготовление см. п.2.10.2.
Кислота сульфосалициловая, раствор с массовой концентрацией 100 г/дм3.
3.5.3 Проведение анализа
Исследуемый образец взвешивают на аналитических весах с точностью до четвертого знака и растворяют в выбранном электролите.
Силу тока рассчитывают в зависимости от площади поверхности исследуемого образца.
По окончании электролиза выключают ток, образец вынимают из коллодиевого мешочка, опускают в стакан с водой. В другой стакан сливают электролит из мешочка с остатками осадка. Осадок с образца тщательно счищают стеклянной палочкой с резиновым наконечником, последние остатки осадка снимают кусочками влажной фильтровальной бумаги. Электролит с остатками осадка и раствор с осадком поочередно переносят на двойной фильтр "синяя лента" диаметром 11 мм. Осадок на фильтре промывают водой до отрицательной реакции на ион хлора (проба с азотнокислым серебром). Фильтр с осадком переносят в фарфоровый или платиновый тигель и высушивают. Фильтр озоляют, осадок прокаливают в муфельной печи при температуре 700 °С и сплавляют с 2-3 г пиросернокислого калия. После охлаждения тигель с расплавом помещают в стакан вместимостью 200 см3 и растворяют расплав в 40 см3 серной кислоты 1:4 при нагревании. Полученный раствор охлаждают, переносят в мерную колбу вместимостью 100-200 см3, доводят до метки водой и перемешивают. Из полученного раствора (А) отбирают аликвотные части.
Образец после электролиза промывают спиртом, высушивают на воздухе и взвешивают на аналитических весах. Разность масс образца до и после электролиза является массой навески растворенного металла, и ее значение используют при расчетах процентного содержания элементов в анодном осадке.
3.5.4 Определение хрома
3.5.4.1 Проведение анализа
Аликвотную часть раствора А в количестве 20-40 см3 (в зависимости от содержания хрома) переносят в коническую колбу вместимостью 250 см3, прибавляют 40-50 см3 смеси кислот и воду, чтобы общий объем раствора в колбе составил 100 см3. Содержание колбы нагревают, прибавляют 10 см3 раствора азотнокислого серебра с массовой концентрацией 2 г/дм3, 15-25 см3 раствора аммония надсернокислого с массовой концентрацией 200 г/дм3 и кипятят 20 мин до полного разрушения избытка надсернокислого аммония. Появление малиновой окраски от марганцовой кислоты указывает на полное окисление трехвалентного хрома в шестивалентный. О разложении надсернокислого аммония судят по исчезновению мелких пузырьков при кипении раствора.
К раствору приливают 5 см3 раствора хлористого натрия с массовой концентрацией 50 г/дм3, нагревают до исчезновения окраски марганцовой кислоты и коагуляции осадка хлорида серебра, охлаждают до комнатной температуры, прибавляют из бюретки точно отмеренное количество титрованного раствора соли Мора, необходимое для полного восстановления хромовой кислоты, о чем судят по изменению цвета от желтого до зеленовато-голубого, и еще 5-10 см3. Избыток соли Мора оттитровывают раствором марганцовокислого калия до розовой окраски, не исчезающей в течение 1 мин.
3.5.4.2 Расчет результатов анализа
Содержание хрома (Cr) в процентах вычисляют по формуле
, (25)
где T - титр раствора марганцовокислого калия, г/см3;
K - соотношение титрованных растворов марганцовокислого калия и соли Мopa;
V2 - объем раствора марганцовокислого калия с массовой концентрацией эквивалента 0,02 моль/дм3, израсходованный на титрование избытка соли Мopa, см3;
V1 - объем раствора соли Мopa с массовой концентрацией эквивалента 0,02 моль/дм3, взятой для титрования, см3;
m - навеска растворенной стали, соответствующая взятой аликвотной части раствора А, г.
3.5.5 Определение хрома фотоколориметрическим методом с дифенилкарбазидом
3.5.5.1 Сущность метода
Дифенилкарбазид в сернокислых растворах восстанавливает шестивалентный хром до двухвалентного состояния. Образуется растворимое соединение, окрашенное в красно-фиолетовый цвет.
3.5.5.2 Реактивы
Смесь кислот; 420 см3 воды, 40 см3 серной кислоты пл. 1,84 охлаждают и прибавляют 40 см3 фосфорной кислоты пл. 1,7.
Серебро азотнокислое, раствор с массовой концентрацией 2 г/дм3.
Дифенилкарбазид, раствор с массовой концентрацией 1,5 г/дм3 в этиловом спирте. Для приготовления 100 г раствора - 0,15 г дифенилкарбазида растворяют в 100 г этилового спирта при нагревании до 40-50 °С. Раствор пригоден в течение 1-2 дней.
Аммоний надсернокислый, раствор с массовой концентрацией 100 г/дм3.
Двухромовокислый калий, стандартный раствор: 0,1132 г перекристаллизованного и высушенного при температуре 130-150 °С двухромовокислого калия растворяют в 200 см3 воды в мерной колбе вместимостью 2 дм3 и доводят до метки водой. В 1 см3 раствора содержится 0,00002 г хрома.
3.5.5.3 Проведение анализа
В мерную колбу вместимостью 100 см3 отбирают пипеткой аликвотную часть раствора - 5-10 см3, прибавляют 10 см3 смеси кислот, 2 см3 раствора азотнокислого серебра с массовой концентрацией 2 г/дм3, 5 см3 раствора аммония надсернокислого с массовой концентрацией 100 г/дм3, осторожно кипятят в течение 3-5 мин до полного окисления хрома (появления малинового окрашивания). Раствор охлаждают, доводят до метки водой, перемешивают. Из этой колбы отбирают в зависимости от содержания хрома 5-10 см3 раствора в другую мерную колбу вместимостью 100 см3, прибавляют 40 см3 воды, 10 см3 спиртового раствора дифенилкарбазида с массовой концентрацией 1,5 г/дм3 и выдерживают 3 мин для развития окраски, затем доливают до метки водой и колориметрируют на фотоколориметре с зеленым светофильтром в кювете рабочей длиной 50 мм.
Для холостого опыта используют воду.
Содержание хрома рассчитывают по калибровочному графику.
3.5.5.4 Расчет результатов анализа
В мерные колбы вместимостью по 100 см3 отбирают из микробюретки стандартный раствор бихромата калия 1; 2; 3... 10 см3, приливают 10 см3 смеси кислот (см. п.2.12.2), 2 см3 раствора азотнокислого серебра с массовой концентрацией 2 г/дм3, 5 см3 раствора аммония надсернокислого с массовой концентрацией 100 г/дм3, осторожно кипятят в течение 3-5 мин до полного окисления хрома. Растворы в колбах охлаждают, доводят до метки водой и перемешивают. Из этих колб отбирают по 10 см3 раствора в другие мерные колбы вместимостью по 100 см3, приливают в них по 10 см3 спиртового раствора дифенилкарбазида с массовой концентрацией 1,5 г/дм3, через 3 мин доливают до метки водой и колориметрируют с зеленым светофильтром в кювете рабочей длиной 50 мм.
3.5.6 Определение ванадия
3.5.6.1 Проведение анализа
Аликвотную часть раствора А, соответствующую навеске растворенной стали не менее 1 г, помещают в коническую колбу вместимостью 250 см3, приливают 50 см3 смеси кислот (см. п.3.5.5.2) и 100 см3 воды до общего объема 150 см3. Для окисления ванадия прибавляют по каплям раствор марганцовокислого калия с массовой концентрацией 5 г/дм3 до появления розовой окраски, не исчезающей в течение 1-2 мин. Марганцовокислый калий восстанавливают одной-двумя каплями щавелевой кислоты, избегая ее избытка, и прибавляют две-три капли раствора фенилантраниловой кислоты с массовой концентрацией 2 г/дм3. Раствор окрашивается в темно-вишневый цвет, его выдерживают 1-2 мин и титруют ванадий растворов соли Мора с массовой концентрацией эквивалента 0,02 моль/дм3.
3.5.6.2 Расчет результатов анализа
Содержание ванадия (V) в процентах вычисляют по формуле
, (26)
где T - титр раствора соли Мора, г/см3;
V - объем раствора соли Мора, израсходованного на титрование, см3;
m - навеска растворенной стали, соответствующая взятой аликвотной части раствора, А, г.
3.5.7 Определение молибдена
Аликвотную часть раствора А в количестве 0,5-10 см3 (в зависимости от содержания молибдена) помещают в мерную колбу вместимостью 100 см3, прибавляют 50 см3 смеси кислот (см. п.2.16.2), 1 см3 раствора сернокислой меди с массовой концентрацией 10 г/дм3, 10 см3 раствора тиомочевины с массовой концентрацией 100 г/дм3, перемешивают. Через 5 мин прибавляют раствор роданистого аммония с массовой концентрацией 250 г/дм3, выдерживают 5-7 мин, доводят до метки водой, перемешивают и через 30 мин колориметрируют с синим светофильтром в кювете рабочей длиной 30 мм.
Для холостого опыта берут воду с добавлением всех реактивов.
Содержание молибдена рассчитывают по калибровочному графику. Построение калибровочного графика см. п.2.16.5.
3.5.8 Определение марганца (объемный персульфатно-серебряный метод)
3.5.8.1 Проведение анализа
Аликвотную часть раствора А в количестве 15-30 см3 помещают в коническую колбу вместимостью 250 см3, прибавляют 40-50 см3 смеси кислот (см. п.2.10.2) и 100 см3 воды до 150 см3. Раствор нагревают до кипения, приливают 10 см3 раствора азотнокислого серебра с массовой концентрацией 2 г/дм3, 15-20 см3 раствора надсернокислого аммония с массовой концентрацией 200 г/дм3, снова кипятят в течение 0,5-1,0 мин до образования крупных пузырьков по всему объему раствора и затем охлаждают в проточной воде до комнатной температуры.
Охлажденный раствор титруют раствором тиосульфата натрия, с массовой концентрацией 1,24 г/дм3 сначала быстро до перехода фиолетовой окраски в розовую, затем медленно (по каплям) до исчезновения розовой окраски. Появление желто-зеленой окраски указывает на окончание титрования.
3.5.8.2 Расчет результатов анализа
Содержание марганца (Mn) в процентах определяют по формуле
, (27)
где V - объем титрованного раствора тиосульфата натрия, израсходованного на титрование марганцевой кислоты, см3;
T - титр раствора тиосульфата натрия, г/см3;
m - навеска растворенной стали, соответствующая аликвотной части раствора, А, г.
3.5.9 Определение содержания железа (фотоколориметрический метод с сульфосалициловой кислотой)
3.5.9.1 Проведение анализа
Аликвотную часть раствора А в количестве 1-10 см3 переносят в мерную колбу вместимостью 100 см3, прибавляют 20 см3 воды, 10 см3 раствора сульфосалициловой кислоты с массовой концентрацией 100 г/дм3 и 5 см3 раствора аммиака с массовой концентрацией 250 г/дм3, доливают до метки водой, перемешивают и измеряют оптическую плотность с синим светофильтром в кювете рабочей длиной 50 мм.
Для холостого опыта берут воду с добавлением всех реактивов.
3.5.9.2 Приготовление титрованного раствора железа
Спектрально чистое железо в количестве 0,1 г растворяют при нагревании в 30 см3 соляной кислоты 1:1, переносят в мерную колбу вместимостью 1 дм3, разбавляют до метки водой и перемешивают. Титр железа равен 0,0001 г/см3.
3.5.9.3 Расчет результатов анализа
Содержание железа (Fe) в процентах вычисляют по формуле
, (28)
где T - титр стандартного раствора железа, г/см3;
Vст - объем стандартного раствора железа, см3;
Dисл - оптическая плотность исследуемого раствора;
m - навеска растворенной стали, соответствующая взятой аликвотной части раствора А, г.
Примечание. Содержание значительных количеств марганца в анодном осадке может вызвать помутнение растворов сульфосалицилата железа; для предотвращения этого в колориметрируемый раствор добавляют 10 см3 раствора солянокислого гидроксиламина с массовой концентрацией 100 г/дм3.
3.6 ФАЗОВЫЙ АНАЛИЗ АУСТЕНИТНЫХ СТАЛЕЙ
(ЭИ257, ЭИ695, ЭИ756 и др.)
3.6.1 Общие положения
Исследование фазового состава аустенитных сталей методом физико-химического фазового анализа проводят в сочетании с рентгеноструктурным методом анализа фаз, исследованием структуры металлографическим методом с применением световой и электронной микроскопии, что позволяет судить о распределении фаз в сплаве, их дисперсности и тонкой структуре.
Выбор состава электролита и режима электролиза обусловливается термодинамической устойчивостью основы сплава и присутствующих в нем фаз (приложение 2).
Широкое распространение для изолирования фаз Me23C6, MeC, Me(CN), Me7C3, σ получил электролит следующего состава: 300 г/дм3 хлористого калия, 50 г/дм3 аммония лимоннокислого, 50 см3/дм3 соляной кислоты пл. 1,19 при плотности тока 0,3-0,5 А/см2 площади поверхности образца.
Для изолирования γ-фазы на основе Ni3Al или Ni3(AlTi) и других аналогичных им фаз применяют электролит следующего состава: 10 г/дм3 лимонной кислоты, 10 г/дм3 аммония сернокислого при плотности тока 0,07 А/см2 площади поверхности образца. Установку для электролитического выделения фаз, подготовку образцов для анализа, снятие осадков, озоление, сплавление выделенных фаз и растворение расплава ведут по п.3.5.3.
При анализе анодных осадков, выделенных из сплавов, легированных вольфрамом, молибденом и ванадием, расплав обрабатывают следующим образом: охлаждают, растворяют в стакане вместимостью 300-400 см3 в смеси соляной и азотной кислот (60 см3 соляной кислоты 1:1 и 5 см3 азотной кислоты 1:1); раствор выпаривают до пастообразного состояния, осадок осторожно подсушивают. К сухому остатку прибавляют 40 см3 соляной кислоты 1:4, нагревают в течение 1 ч, приливают 100 см3 воды и нагревают еще в течение 1 ч. Выделившийся осадок вольфрамовой кислоты отфильтровывают и промывают раствором соляной кислоты 1:10 до отрицательной реакции на ион двухвалентного никеля. К солянокислому фильтрату прибавляют 15 см3 серной кислоты пл. 1,84, выпаривают до появления белых паров серной кислоты, охлаждают, приливают 150 см3 воды и вновь нагревают до полного растворения солей.
Если при этом выделится дополнительный осадок вольфрамовой кислоты, его отфильтровывают, промывают на фильтре раствором серной кислоты 1:100. Фильтрат после отделения вольфрама переносят в мерную колбу вместимостью 200 см3, разбавляют до метки водой и перемешивают. В этом фильтрате определяют содержание легирующих элементов, входящих в состав анодных осадков. Определение каждого элемента производят из аликвотных частей фильтрата. Хром определяют титриметрическим персульфатно-серебряным методом (см. пп.2.12 или 3.5.4), титан - фотоколориметрическим методом с диантиперилметаном (см. п.2.17), молибден - фотоколориметрическим методом с роданистым аммонием (см. п.2.16), ниобий - фотоколориметрическим методом (см. п.2.21).
В оставшемся осадке можно определить вольфрам гравиметрическим методом, но, так как гидролизом не всегда удается достигнуть полноты выделения вольфрамовой кислоты, дополнительно из аликвотной части фильтрата определяют содержание вольфрама колориметрическим методом.
Практика показала, что для определения содержания вольфрама в анодном осадке лучше отдельно выделить электролитический осадок.
3.6.2 Подготовка анодного осадка для определения вольфрама
Анодный осадок отфильтровывают и отмывают от электролита несколько раз водой. Фильтр с осадком помещают в стакан вместимостью 300 см3 с 20 см3 раствора гидроксида натрия с массовой концентрацией 100 г/дм3 и выдерживают 20 мин, добавляют 20 см3 раствора лимонной кислоты с массовой концентрацией 200 г/дм3, отфильтровывают и отмывают водой от гидроксида натрия - проба с фенолфталеином.
Фильтр с осадком переносят в фарфоровый тигель, осторожно подсушивают, озоляют и сплавляют с пиросернокислым калием.
В стакане вместимостью 250-300 см3 плав выщелачивают водой и для отделения вольфрама от сопутствующих элементов нейтрализуют раствором гидроксида натрия с массовой концентрацией 200 г/дм3 до начала выделения осадка гидрооксидов, который растворяют в нескольких каплях серной кислоты пл. 1,84. Прозрачный раствор нагревают и осторожно при постоянном помешивании переливают в мерную колбу вместимостью 200 см3, содержащую 20 см3 горячего раствора гидроксида натрия с массовой концентрацией 200 г/дм3. Раствор охлаждают, разбавляют водой до метки, перемешивают и отфильтровывают через сухой плотный фильтр в сухой стакан. Из аликвотной части полученного фильтрата определяют содержание вольфрама фотоколориметрическим методом (см. п.2.26).
3.7 ДИФФЕРЕНЦИРОВАННЫЙ ФАЗОВЫЙ АНАЛИЗ СТАЛИ X18Н12Т
В состоянии поставки в стали X18H12T присутствуют карбиды и нитриды титана - TiC, TiN. Длительное старение или эксплуатация при температуре 500-800 °С приводит к образованию в стали σ-фазы состава FeCr, карбида Me23C6, двойного карбида (TiNi)6C.
В структуре стали X18H12T наблюдаются следующие комбинации карбидных и интерметаллидных фаз:
TiC + Me23C6;
TiC + σ-фазы;
TiC + σ-фазы + (TiNi)6C;
TiC + σ-фазы + Me23C6;
TiC + σ-фазы + Me23C6 + (TiNi)6C.
Для определения количества и состава фаз используется метод двух электролитов.
Первый электролит - 300 г/дм3 хлористого калия, 50 см3/дм3 соляной кислоты пл. 1,19, 30 г/дм3 лимонной кислоты; плотность тока 0,7 А/см2 площади поверхности образца, температура электролита - 0-5 °С, продолжительность растворения 10-15 мин. В анодный осадок (1) переходят фазы: TiC, σ-фаза, Me23C6, (TiNi)6C. Анализируя осадок (1), определяют общее количество легирующих элементов в фазах.
Второй электролит - соляная кислота 1:2, 10 г/дм3 лимонной кислоты; плотность тока 0,05-0,08 А/см2 площади поверхности образца, температура электролита 18-20 °С, продолжительность растворения 1,5-2 ч. В анодный осадок (2) переходят только карбидные фазы. σ-фаза не изолируется.
Анализ двух анодных осадков позволит установить количество легирующих элементов в выделенных фазах; количество σ-фазы находят по разности содержания хрома и железа в осадках (1) и (2), количество карбида Me23C6 - по содержанию хрома и железа в осадке (2). Наличие двойного карбида (TiNi)6C - по содержанию никеля и титана в осадке (2).
В анодном осадке, выделенном из стали X18H12T или X18H10T, определяют: хром - титриметрическим персульфатно-серебряным методом фенилантраниловой кислотой (см. п.2.12); железо - фотоколориметрическим методом с сульфосалициловой кислотой (см. п.3.5.9), никель - с диметилглиоксимом (см. п.2.19); титан - с диантиперилметаном (см. п.2.17) или с перекисью водорода.
Для фотометрического метода определения титана с перекисью водорода аликвотную часть раствора в количестве 5-20 см3 переносят в мерную колбу вместимостью 50 см3, прибавляют 20 см3 воды, 1 см3 фосфорной кислоты пл. 1,7, 2 см3 перекиси водорода, доводят объем до метки водой, перемешивают. Растворы надтитановой кислоты нестойки во времени. Фотометрируют с синим светофильтром в кюветах рабочей длиной 30 мм. Нуль гальванометра устанавливают по холостой пробе.
Содержание гитана (Ti) в процентах рассчитывают по стандартному раствору титана методом сравнения.
, (29)
где Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора;
T - титр стандартного раствора, г/см3;
mст - навеска стандартного раствора, г;
ma - навеска растворенной стали, соответствующая взятой аликвотной части, г.
Стандартный раствор титана готовят из двуокиси титана: 0,1663 г двуокиси титана сплавляют в платиновом тигле с 2-3 г пиросернокислого калия. Плав выщелачивают в 100 см3 серной кислоты 1:1, переводят в мерную колбу вместимостью 1 дм3, доводят до метки водой, перемешивают.
Титр титана равен 0,1 мг/см3.
В колбы вместимостью по 50 см3 отбирают по 1-3 см3 стандартного раствора титана. Интервал концентрации титана в объеме 50 см3 составляет 0,125; 0,15; 0,175; 0,3 мг. Оптическую плотность растворов измеряют в кювете рабочей длиной 30 мм, она не должна превышать значения 0,24.
Определение никеля (фотоколориметрический метод с диметилглиоксимом).
Аликвотную часть раствора А в количестве 2-20 см3 (в зависимости от содержания никеля в анодном осадке) переносят в мерною колбу вместимостью 100 см3, куда добавляют при перемешивании реактивы в следующее порядке:
20 см3 воды;
10 см3 раствора сегнетовой соли с массовой концентрацией 200 г/дм3;
10 см3 раствора гидроксида натрия с массовой концентрацией 100 г/дм3;
10 см3 раствора надсернокислого аммония с массовой концентрацией 100 г/дм3;
10 см3 раствора диметилглиоксима с массовой концентрацией 10 г/дм3, приготовленного на растворе гидроксида натрия с массовой концентрацией 50 г/дм3.
После пятиминутной выдержки растворы разбавляют водой до метки, перемешивают и через 5-10 мин колориметрируют с синим светофильтром в кюветах рабочей длиной 30 мм. Лучшая сходимость результатов получается при колориметрировании слабоокрашенных растворов при содержании никеля 0,01-0,05 мг в 100 см3. Нуль гальванометра устанавливают по холостой пробе, для этого к аликвотной части раствора А прибавляют последовательно все реактивы, кроме диметилглиоксима, который заменяют 5 см3 раствора гидроксида натрия с массовой концентрацией 100 г/дм3.
Содержание никеля (Ni) в процентах рассчитывают по стандартному раствору никеля методом сравнения
, (30)
где Dисл - оптическая плотность исследуемого раствора;
Dст - оптическая плотность стандартного раствора;
T - титр стандартного раствора никеля, г/см3;
mст - навеска стандартного раствора, соответствующая аликвотной части, г;
ma - навеска растворенной стали, соответствующая взятой аликвотной части, г.
Стандартный раствор никеля готовят следующим образом: 0,1 г металлического никеля высокой чистоты растворяют в 20 см3 соляной кислоты 1:4, прибавляют три-пять капель азотной кислоты пл. 1,40. Раствор кипятят до удаления оксидов азота, охлаждают и переносят в мерную колбу вместимостью 1 дм3, доливают водой до метки, перемешивают; 1 см3 такого раствора содержит 0,00001 г никеля. Содержание никеля можно также определять по калибровочному графику. Для его построения в мерные колбы вместимостью до 100 см3 приливают из микробюретки 0,5; 1,0; 1,5... 8,0 см3 стандартного раствора, содержащего 0,00001 г никеля в 1 см3.
В каждую колбу при перемешивании прибавляют реактивы, указанные в ходе анализа. Через 5 мин растворы в колбах доливают до метки водой, перемешивают и колориметрируют с синим светофильтром в кюветах рабочей длиной 30 мм.
Примечание. В случае появления в колориметрируемых растворах диметилглиоксимата никеля мути или осадка перед доведением раствора до метки водой прибавляют по каплям раствор гидроксида натрия с массовой концентрацией 300 г/дм3 до полного осветления раствора.
3.8 ФАЗОВЫЙ АНАЛИЗ СТАЛЕЙ 12Х1МФ И 15Х1М1Ф МЕТОДОМ ХИМИЧЕСКОГО РАСТВОРЕНИЯ
3.8.1 Реактивы и приспособления
Кислота серная, 1:6.
Кислота лимонная, раствор с массовой концентрацией 100 г/дм3.
Смесь кислот; 750 см3 воды и 200 см3 серной кислоты пл. 1,84 охлаждают, прибавляют 50 см3 соляной кислоты пл. 1,19.
Медь сернокислая, раствор с массовой концентрацией 20 г/дм3.
Тиомочевина, раствор с массовой концентрацией 100 г/дм3.
Калий или аммоний роданистый, раствор с массовой концентрацией 250 г/дм3.
Калий пиросернокислый.
Шабер.
Магнит для сбора стружки.
Пробирки для отбора проб емкостью по 20 см3.
Химические стаканы вместимостью по 50 см3.
Воронки диаметром 30 мм.
Фильтры "синяя лента".
Колбы мерные вместимостью 25 см3, 50 см3 и 100 см3.
Тигли фарфоровые № 5.
3.8.2 Отбор пробы для анализа
С анализируемого участка отбирают пробу стали в виде стружки. Предварительно шлифовальной машинкой зачищают исследуемое место размером 100х40 мм, обезжиривают спиртом или эфиром. Шабером снимают стружку в количестве 0,4-0,6 г и собирают ее магнитом в пробирку или мешочек из лощеной бумаги или кальки. Записывают место отбора пробы.
Отбор пробы можно производить с помощи пневмосверлилки, на которую надевается специальная фреза.
3.8.3 Определение молибдена
Навеску (стружку) массой 0,2 г растворяют без нагревания в химическом стакане вместимостью 50 см3 в 20 см3 серной кислоты 1:6. После растворения стружки карбидный осадок фильтруют через двойной фильтр "синяя лента" диаметром 9 см. Промывают два раза водой, затем два-три раза раствором лимонной кислоты с массовой концентрацией 10 г/дм3 и снова водой.
Фильтр с осадком помещают в фарфоровый тигель № 5, сжигают в муфельной печи при температуре 500-600 °С и сплавляют с 1-2 г пиросернокислого калия. При полном сплавлении осадка плав становится прозрачным. Плав выщелачивают водой и переводят в мерную колбу вместимостью 25 см3. Аликвотную часть раствора от 1 до 15 см3 (в зависимости от содержания молибдена в карбидном осадке) переносят в мерную колбу вместимостью 100 см3, прибавляют 40 см3 смеси кислот, 2 см3 раствора сернокислой меди с массовой концентрацией 20 г/дм3, 10 см3 раствора тиомочевины с массовой концентрацией 100 г/дм3 и через 2-3 мин 4 см3 раствора роданистого аммония с массовой концентрацией 250 г/дм3. Через 30 мин измеряют оптическую плотность тока с синим светофильтром в кювете рабочей длиной 30 мм.
Содержание молибдена рассчитывают по калибровочному графику, построенному по стандартному образцу с содержанием молибдена 0,5% (см. п.2.16).
Для определения содержания молибдена в твердом растворе надо взять соответствующую аликвотную часть из фильтрата после отделения карбидного осадка и сделать анализ на содержание молибдена.
3.8.4 Определение хрома
Для выделения карбидов хрома 0,2 г отобранной пробы (стружки) растворяют в 20 см3 азотной кислоты 1:4, охлажденной до температуры 10-15 °С. Осадок отфильтровывают на фильтр "синяя лента", если остается нерастворенная стружка, ее помещают в стакан и наливают свежую порцию азотной кислоты. После полного растворения стружки осадок карбидов фильтруют через тот же фильтр. Фильтр с осадком помещают в фарфоровый тигель, подсушивают, озоляют и сплавляют с пиросернокислым калием; плав выщелачивают в 20 см3 серной кислоты 1:4. Содержание хрома определяют персульфатно-серебряным методом из всей навески. Этот метод пригоден для определения хрома в стали марки 12Х1МФ. Из стали марки 15X1M1Ф данным методом можно выделить только 60-70% карбидов хрома.
Расчет результатов анализа см. п.3.5.4.2.
4 ПОРЯДОК УЧЕТА РЕЗУЛЬТАТОВ АНАЛИЗА И ХРАНЕНИЯ ПРОБ
4.1 Образец, поступающий в лабораторию на анализ, регистрируется в книге учета раздельно на химический и физико-химический фазовый анализы по форме (таблица 1).
Таблица 1 - Форма регистрационного журнала для образца
Номер п/п дата | Клеймо | Наименование ТЭЦ или фамилия и инициалы заказчика | Место отбора пробы, дата отбора | Марка стали по сертификату | Определяемые элементы, % | ||||||||||
С | Mn | Si | S | P | Cr | Mо | V | Ti | Ni | и т.д. | |||||
(При фазовом анализе определяются следующие элементы: Мо, Cr, V, W, Ti, Mn, Fe …).
Для записей в ходе исследования ведут рабочую тетрадь, в которую заносятся: наименование ТЭС или фамилия, инициалы заказчика, регистрационный номер образца в книге учета, элементы, подлежащие определению.
В рабочей тетради для фазового анализа записывают также условия электролиза: состав электролита, плотность тока, массу растворенной стали, объем растворения, определяемые элементы, аликвотные части, взятые для анализа.
Запись хода и результатов анализа подписывает лицо, производившее анализ.
Пробы металла после выполнения исследования хранятся в лаборатории в течение 6 мес. со дня поступления.
Оборудование, необходимое для химических лабораторий служб металлов, приведено в приложении 3.
4.2 Заказчику выдают заключение о результатах анализа, содержащее следующие сведения:
а) наименование ТЭС (или иного заказчика);
б) марка стали;
в) клеймо;
г) место отбора пробы, дата отбора;
д) обозначение и наименование методик (методов), по которым был проведен анализ;
е) отклонения, допущенные или замеченные в ходе анализа;
ж) результат анализа по каждому элементу.
Заключение подписывают заведующий лабораторией и исполнитель анализа.
Приложение 1
Справочное
ДОПУСТИМЫЕ РАСХОЖДЕНИЯ РЕЗУЛЬТАТОВ ХИМИЧЕСКОГО АНАЛИЗА СТАЛИ
Определяемый элемент | Метод анализа | Массовая доля элемента, % | Допускаемые расхождения между результатами двух измерений, % |
1 | 2 | 3 | 4 |
Углерод, графит | Газообъемный, кулонометрический | от 0,05 до 0,10 вкл. | 0,010 |
-"- 0,10 -"- 0,20 -"- | 0,015 | ||
-"- 0,20 -"- 0,50 -"- | 0,020 | ||
Сера | Сжигание в токе кислорода | от 0,01 до 0,025 вкл. | 0,004 |
-"- 0,025 " 0,05 " | 0,006 | ||
" 0,05 " 0,10 " | 0,008 | ||
Фосфор | Фотоколориметрический | от 0,01 до 0,02 вкл. | 0,0025 |
" 0,02 " 0,05 " | 0,004 | ||
" 0,05 " 0,10 " | 0,006 | ||
Марганец | Титриметрический персульфатный | от 0,10 до 0,20 вкл. | 0,01 |
" 0,20 " 0,40 " | 0,02 | ||
" 0,40 " 1,00 " | 0,03 | ||
" 1,00 " 1,50 " | 0,04 | ||
Кремний | Гравиметрический, фотоколориметрический | от 0,05 до 0,10 вкл. | 0,01 |
" 0,10 " 0,20 " | 0,02 | ||
" 0,20 " 0,50 " | 0,03 | ||
" 0,50 " 1,00 " | 0,04 | ||
Молибден | Фотоколориметрический | от 0,10 до 0,25 вкл. | 0,03 |
" 0,25 " 0,60 " | 0,04 | ||
" 0,60 " 1,00 " | 0,05 | ||
" 1,00 " 2,00 " | 0,07 | ||
Хром | Фотоколориметрический | от 0,01 до 0,025 вкл. | 0,005 |
" 0,025 " 0,05 " | 0,007 | ||
" 0,05 " 0,10 " | 0,010 | ||
" 0,10 " 0,20 " | 0,015 | ||
Хром | Титриметрический персульфатный | от 0,20 до 0,50 вкл. | 0,02 |
" 0,50 " 1,0 " | 0,03 | ||
" 1,0 " 2,0 " | 0,05 | ||
" 2,0 " 5,0 " | 0,07 | ||
" 5,0 " 10,0 " | 0,10 | ||
Никель | Фотоколориметрический | от 0,08 до 0,15 | 0,015 |
св. 0,15 до 0,3 | 0,02 | ||
св. 0,3 до 0,5 | 0,03 | ||
Никель | Гравиметрический | от 0,50 до 1,0 | 0,05 |
св. 1,0 до 2,0 | 0,06 | ||
св. 2,0 до 4,0 | 0,08 | ||
св. 4,0 до 8,0 | 0,12 | ||
св. 8,0 до 15,0 | 0,16 | ||
св. 15,0 до 25,0 | 0,20 | ||
Ванадий | Титриметрический | от 0,10 до 0,25 | 0,03 |
св. 0,25 до 0,5 | 0,04 | ||
св. 0,5 до 1,0 | 0,06 | ||
Вольфрам | Фотоколориметрический | от 0,10 до 0,4 | 0,02 |
св. 0,4 до 0,5 | 0,04 | ||
св. 0,5 до 1,0 | 0,06 | ||
св. 1,0 до 2,0 | 0,1 | ||
Титан | Фотоколориметрический | от 0,05 до 0,1 | 0,02 |
св. 0,1 до 0,2 | 0,03 | ||
св. 0,2 до 0,5 | 0,04 | ||
св. 0,5 до 1,0 | 0,07 | ||
Ниобий | Фотоколориметрический | от 0,09 до 0,1 | 0,01 |
св. 0,1 до 0,5 | 0,03 | ||
св. 0,5 до 1,0 | 0,05 | ||
св. 1,0 до 3,0 | 0,08 | ||
Медь | Фотоколориметрический | от 0,01 до 0,02 | 0,005 |
св. 0,02 до 0,05 | 0,008 | ||
св. 0,05 до 0,1 | 0,01 | ||
св. 0,1 до 0,25 | 0,02 |
Приложение 2
Рекомендуемое
СОСТАВЫ ЭЛЕКТРОЛИТОВ, ПРИМЕНЯЕМЫЕ ДЛЯ ИЗОЛИРОВАНИЯ ФАЗ ИЗ СТАЛЕЙ С КАРБИДНЫМ И ИНТЕРМЕТАЛЛИДНЫМ УПРОЧНЕНИЯМИ
Состав электролита | Режим электролиза | Класс стали | Изолируемые фазы | |
Плотность тока, А/см2 | Температура электролита, °С | |||
1 | 2 | 3 | 4 | 5 |
Калий хлористый, раствор с массовой концентрацией 75 г/дм3; кислота лимонная с массовой концентрацией 50 г/дм3 | 0,02-0,05 | от -5 до 0 | Углеродистая, низколегированная, перлитная и мартенситная | Me3C, Me7C3, Me23С6, МеС |
Калий бромистый, раствор с массовой концентрацией эквивалента 0,2 моль/дм3; натрий лимоннокислый, раствор с массовой концентрацией эквивалента 0,5 моль/дм3; аскорбиновая кислота с массовой концентрацией 5 г/дм3 | - | - | Низколегированная, углеродистая | Me3C, Me7C3, VС |
Натрий лимоннокислый, раствор с массовой концентрацией 50-100 г/дм3; калий хлористый или йодистый, раствор с массовой концентрацией 20 г/дм3 | - | - | Сталь, содержащая менее 5% хрома | Карбиды, нитриды, интерметаллиды |
Натрий лимоннокислый, раствор с массовой концентрацией 50 г/дм3; калий бромистый, р-р с массовой концентрацией 12 г/дм3; дибензилсульфоксид, раствор с массовой концентрацией 0,25 г/дм3; калий йодистый, раствор с массовой концентрацией 6 г/дм3; натрий фтористый, раствор с массовой концентрацией 1,1 г/дм3 | - | - | Графитосодержащая, углеродистая и легированные чугуны | Карбиды, нитриды, графит, окислы, сульфиды |
Аммоний хлористый, раствор с массовой концентрацией 300 г/дм3; сахароза, раствор с массовой концентрацией 10 г/дм3 | 0,03 | +10 | Железо-алюминий- кремний-углерод | ε-фаза Fe3AlCx |
Аммоний роданистый, раствор с массовой концентрацией 10 г/дм3; магний уксуснокислый, раствор с массовой концентрацией 5 г/дм3 | 0,02 | -5÷+10 | 9Г28Ю8М | К-фаза (Fe1Mn)3AlCx |
Калий хлористый, раствор с массовой концентрацией 75 г/дм3; кислота соляная, раствор с массовой концентрацией эквивалента 0,02 моль/дм3; глицерин, раствор с массовой концентрацией 100 г/дм3 | 0,01 | -5÷-3 | Аустенитная , высокомарганцовистая | Me3C, Me7C3 |
Кислота соляная, раствор с объемной концентрацией 20 cм3/дм3; глицерин, спиртовой раствор с массовой концентрацией 50 г/дм3 | 0,05 | - | Низколегированная марганцовистая | V(C,N), VC α-феррит |
Калий хлористый, раствор с массовой концентрацией 75 г/дм3; тиомочевина, раствор с массовой концентрацией 2-3 г/дм3; кислота соляная, раствор с объемной концентрацией 50-500 см3/дм3 | 0,02 | +20 | Мартенситная с 12% Cr, аустенитная с карбидным упрочнением | Me23C6, Mе2(C,N), Me23C, Me7C3, MeCХ, Me(C,N), |
Натрий хлористый, раствор с массовой концентрацией 150 г/дм3; кислота винная, раствор с массовой концентрацией 25 г/дм3 | 0,07-1,2 | +20 | Мартенситная с 12% Cr, аустенитная с карбидным упрочнением | МеС, Me7C3, Me6С, σ, Me(C,N), |
Натрий лимоннокислый, раствор с массовой концентрацией 200 г/дм3; натрий хлористый, раствор с массовой концентрацией 30 г/дм3; кислота аскорбиновая, раствор с массовой, концентрацией 10 г/дм3 | - | - | Аустенитная | Карбиды, окислы |
Натрий фтористый, раствор с массовой концентрацией 20-30 г/дм3; кислота лимонная, раствор с массовой концентрацией 10 г/дм3; кислота соляная, раствор с объемной концентрацией 50-100 см3/дм3 | 0,02 | +20 | Нержавеющая | МеС, Me7C3, Me6С, Me23C6, NiBe, Ме3B2 |
Кислота соляная, спиртовой раствор с объемной концентрацией 50 см3/дм3 | 0,02-0,10 | +20 | Высоколегированная: мартенситная с 12% Cr, штамповая, мартенсито-стареющая, аустенитная с карбидным упрочнением | Me23C6, Me7C3, Ме6С, Me2(C,N), MeС, |
Кислота соляная, раствор с объемной концентрацией 250 см3/дм3; кислота лимонная, раствор с массовой концентрацией 30 г/дм3 | 0,02 | +20 | Сталь переходного класса и высокохромистая | Me2C, Ме(С, N), Me23C6, σ, χ |
Калий хлористый, раствор с массовой концентрацией 300 г/дм3; кислота лимонная, раствор с массовой концентрацией 20-50 г/дм3; кислота соляная, раствор с объемной концентрацией 50 см3/дм3 | 0,1-1,0 | 0 | Нержавеющая коррозионно-стойкая | Me23C6, MeС, Me(C, N), Me7C3, |
Кислота соляная, раствор с объемной концентрацией 50 см3/дм3; глицерин в метиловом спирте, раствор с массовой концентрацией 100 г/дм3 | 0,07 | -5÷-7 | Все классы сталей | МеС, Me7C3, Me23C6, TiB2, |
Соляная кислота, раствор с объемной концентрацией 30-100 см3/дм3 | 0,02-0,05 | 20 | Быстрорежущая | Ме6С, Me23C6, МеС |
Литий хлористый, раствор с массовой концентрацией 100-400 г/дм3; кислота лимонная, раствор с массовой концентрацией 30 г/дм3; кислота соляная, раствор с объемной концентрацией 100 см3/дм3 | 0,02 | -1÷-18 | Быстрорежущая | МеС, Me7C3, Ме6С, Me23C6 и др. |
Кислота щавелевая, раствор с массовой концентрацией 5 г/дм3; кислота соляная, раствор с объемной концентрацией 200 см3/дм3 | 0,05 | 20 | Нержавеющая, коррозионно-стойкая | МеС, Me(C, N), Me23C6, σ, |
Аммоний сернокислый, раствор с массовой концентрацией 10 г/дм3; кислота лимонная, раствор с массовой концентрацией 10-40 г/дм3 | 0,01-0,08 | +5÷+20 | Аустенитная хромоникель-титановая с интерметаллидным упрочнением | Ni3Al, Ni3(Al, Ti), β-Ni3Ti, Ni3Nb, MeC, Me23C6, Me7(W, Mo)6 |
Аммоний сернокислый, раствор с массовой концентрацией 5 г/дм3; кислота лимонная, раствор с массовой концентрацией 35 г/дм3; кислота азотная, раствор с объемной концентрацией 15 см3/дм3 | 0,01-0,08 | +5÷+20 | То же | То же |
Кислота фосфорная, раствор с объемной концентрацией 100-200 см3/дм3 | 0,01-0,08 | +5÷+20 | Аустенитная хромоникель-титановая с интерметаллидным упрочнением | |
Медь сернокислая, раствор с массовой концентрацией 50 г/дм3; натрий лимоннокислый, раствор с массовой концентрацией 30-80 г/дм3; спирт метиловый, раствор с объемной концентрацией 100 см3/дм3 | 0,01-0,08 | +1÷+15 | -"- | -"- |
Железо сернокислое, раствор с массовой концентрацией 30 г/дм3; натрий хлористый, раствор с массовой концентрацией 35 г/дм3; кислота серная, раствор с объемной концентрацией 50 см3/дм3 | 0,05 | +20 | Аустенитная хромоникель-титановая с интерметаллидным упрочнением | Ni3Al, Me7С3, Ni3(Al,Ti), MeC, Ме6С, Me23C6 |
Калий роданистый, раствор с массовой концентрацией 2-3 г/дм3; кислота серная, раствор с объемной концентрацией 50 см3/дм3; кислота лимонная, раствор с массовой концентрацией 1 г/дм3 | 0,01-0,05 | +0÷+10 | То же | Ni3(Al,Ti), Ni3Ti, MeC, Me23C6 |
Приложение 3
Рекомендуемое
ОБОРУДОВАНИЕ, НЕОБХОДИМОЕ ДЛЯ ХИМИЧЕСКИХ ЛАБОРАТОРИЙ СЛУЖБ МЕТАЛЛОВ
Для выполнения аналитических работ по методикам, включенным в данные Методические указания, необходимо следующие оборудование и приборы:
Вытяжной шкаф.
Лабораторные столы.
Столы с амортизаторами для аналитических весов или полка на кронштейнах, вмонтированных в капитальную стену.
Весы аналитические любого типа с точностью взвешивания до 0,0002 г и пределом взвешивания 200 г.
Весы технические с пределом взвешивания 1 кг.
Фотоэлектроколориметры ФЭК-М, ФЭК-Н-56 или КФК-2.
Муфельная лабораторная электропечь типа СНОЛ.
Трубчатая печь с карборундовыми нагревателями и терморегулятором.
Аппарат для газообъемного определения углерода газоанализатор ГОУ-1.
Экспресс-анализатор на углерод типа АН-7529.
Штативы лабораторные (школьные).
Сушильный шкаф типа СНОЛ.
Аппарат для перегонки воды 795АА-1.
Холодильник бытовой.
Электроплиты с закрытой спиралью и регулятором температуры нагрева.
Выпрямители ВСА-6А, ВСА-4А и др.
Амперметры на 1-3 А и на 10-30 А.
Реостаты.
Центрифуга ЦЛН-2.
Приложение 4
Справочное
Перечень нормативных документов, на которые даны ссылки в РД 34.17.414-95
Обозначение стандарта | Наименование стандарта | Номер пункта, приложения, таблицы, на которые даны ссылки |
1 | 2 | 3 |
ГОСТ 7122-81 | Швы сварные и металл наплавленный. Методы отбора проб для определения химического состава | 2.1 |
ГОСТ 7565-81 | Чугун, сталь и сплавы. Метод отбора проб для химического состава | 2.1 |
ГОСТ 12345-88 | Стали легированные и высоколегированные. Методы определения серы | 2.5 |
ГОСТ 12346-78 | Стали легированные и высоколегированные. Методы определения кремния | 2.6 |
ГОСТ 12347-77 | Стали легированные и высоколегированные. Методы определения фосфора | |
ГОСТ 12348-78 | Стали легированные и высоколегированные. Методы определения марганца | 2.11 |
ГОСТ 12349-83 | Стали легированные и высоколегированные. Методы определения вольфрама | 2.20 |
ГОСТ 12350-78 | Стали легированные и высоколегированные. Методы определения хрома | 2.13 |
ГОСТ 12351-81 | Стали легированные и высоколегированные. Методы определения ванадия | 2.14 |
ГОСТ 12352-81 | Стали легированные и высоколегированные. Методы определения никеля | 2.18, 2.19 |
ГОСТ 12354-81 | Стали легированные и высоколегированные. Методы определения молибдена | 2.16 |
ГОСТ 12356-81 | Стали легированные и высоколегированные. Метод определения титана | 2.17 |
ГОСТ 22536.1-88 | Сталь углеродистая и чугун нелегированный. Методы определения общего углерода и графита | 2.2, 2.3, 2.4 |
ГОСТ 22536.3-88 | Сталь углеродистая и чугун нелегированный. Методы определения фосфора | 2.8 |
ГОСТ 22536.4-88 | Сталь углеродистая и чугун нелегированный. Методы определения кремния | 2.7 |
ГОСТ 22536.5-87 | Сталь углеродистая и чугун нелегированный. Методы определения марганца | 2.10 |
ГОСТ 22536.8-87 | Сталь углеродистая и чугун нелегированный. Методы определения меди | 2.22 |
Ключевые слова: энергетика, тепловые электростанции, металлоборудования, анализ, химический, физико-химический
ОГЛАВЛЕНИЕ
1 Общие требования к подготовке и проведению анализов стали
2 Химический анализ сталей
2.1 Отбор проб стали для анализа (по гост 7565 и гост 7122)
2.2 Определение общего углерода
2.3 Определение общего углерода
2.4 Определение свободного углерода
2.5 Определение серы йодометрическим методом
2.6 Определение кремния гравиметрическим методом
2.7 Определение кремния фотоколориметрическим методом
2.8 Определение фосфора фотоколориметрическим методом в углеродистых и низколегированных сталях
2.9 Определение фосфора фотоколориметрическим методом в сталях с содержанием титана, ниобия, вольфрама
2.10 Определение марганца в сталях с содержанием хрома менее 2% и кобальта менее 0,1%
2.11 Определение марганца в сталях с содержанием хрома более 2%
2.12 Определение хрома в сталях, не содержащих ванадий
2.13 Определение хрома в углеродистых солях и сталях, содержащих ванадий
2.14 Определение ванадия титриметрическим методом с фенилантраниловой кислотой
2.15 Определение марганца, хрома и ванадия из одной навески
2.16 Определение молибдена фотоколориметрическим методом
2.17 Определение титана фотоколориметрическим методом с диантиперилметаном
2.18 Определение никеля гравиметрическим методом при его содержании в стали 0,6-30%
2.19 Определение никеля фотоколориметрическим методом
2.20 Определение вольфрама фотоколориметрическим методом
2.21 Определение ниобия фотоколориметрическим методом с цианформазаном-2
2.22 Определение меди фотоколориметрическим методом
3 Фазовый анализ энергетических сталей
3.1 Общие положения
3.2 Подготовка образцов для фазового ahaлизa
3.3 Изготовление коллодиевого мешочка
3.4 Электролитический метод фазового анализа с изготовлением специального образца
3.5 Фазовый анализ перлитных сталей
3.6 Фазовый анализ аустенитных сталей
3.7 Дифференцированный фазовый анализ стали x18н12т
3.8 Фазовый анализ сталей 12х1мф и 15х1м1ф методом химического растворения
4 Порядок учета результатов анализа и хранения проб
Приложение 1 Допустимые расхождения результатов химического анализа стали
Приложение 2 Составы электролитов, применяемые для изолирования фаз из сталей с карбидным и интерметаллидным упрочнениями
Приложение 3 Оборудование, необходимое для химических лабораторий служб металлов
Приложение 4 Перечень нормативных документов, на которые даны ссылки в РД 34.17.414-95